幽门螺杆菌感染性慢性胃炎模型小鼠肠道各区域的菌群分布特征及其机制
2023-05-06覃艳春黄衍强黄干荣唐华英戴园园
覃艳春, 黄衍强, 陆 钢, 黄干荣, 唐华英, 戴园园
(1.右江民族医学院基础医学院病原生物学与免疫学教研室,广西 百色 533000;2.右江民族医学院附属医院烧伤整形与创面修复外科,广西 百色 533000)
幽门螺杆菌(Helicobacter Pylori,Hp)是人类胃中定植的病原体,可诱发消化性溃疡等胃肠道疾病[1],引起上腹疼痛、恶心和嗳气等一系列症状,Hp感染介导机体免疫应答,同时促进胃肠黏膜的自噬,进而影响肠道菌群[2]。肠道菌群在人体的代谢、生理和免疫系统中发挥重要作用[3-6],肠道微生物与肿瘤、糖尿病、帕金森病、神经系统疾病和自闭症谱系障碍等疾病紧密相关[7-11]。随着测序技术的发展,国内外学者对Hp 感染与胃肠道微生物群之间关系进行一系列的探索,但多数集中在胃菌群和粪便标本上[12-14],且尚不清楚粪便样本在多大程度上反映其他肠道区域的微生物组,Hp 感染后肠道各区域菌群的特征迄今尚未见报道。本研究采用16SrRNA 高通量测序技术,探讨Hp 感染性慢性胃炎模型小鼠十二指肠、空肠和结肠肠道菌群种类、特征及菌群差异,为基于肠道菌群根除 Hp感染以预防胃癌提供新思路。
1 材料与方法
1.1 实验动物、主要试剂和仪器30 只6~8 周龄SPF 级C57BL/6 小鼠,体质量(20.0±2.0)g,购自广东维通利华实验动物技术有限公司,动物生产许可证号:SCXK(粤)2022-0063。DNA 提取试剂盒(美国OMEGA 公司,货号:M5635-02),Qubit4.0 DNA 检测试剂盒(美国ThermoFisher 公司,货号:Q32854),PCR 扩增试剂盒(中国Yeasen 公司,货号:10105ES03),DNA 分选磁珠(中国Yeasen 公司,货号:12601ES56),HE 染色液套装(中国Servicebio 公司,货号:G1003)。病理切片机(上海徕卡仪器有限公司,型号:RM2016),正置光学显微镜(日本尼康公司,型号:Nikon Eclipse E100),台式离心机(美国Thermo Fisher 公司,型号:Pico-21),混匀型干式恒温器(深圳拓能达科技有限公司,型号:TND03-H-H),凝胶成像系统(上海复日科技有限公司,型号:FR-1000),QubitR 4.0 荧光计(美国ThermoFisher 公司,型号:Q33226),PCR 仪(北京东胜创新生物科技有限公司,型号:ETC 811)。
1.2 Hp 感染性慢性胃炎小鼠模型的建立根据本校黄衍强博士专利(专利号:ZL2019 1 0245354.5)建模,将30 只6~8 周龄C57BL/6 小鼠随机分为对照组和感染组,每组15 只。感染组小鼠采用临床分离的Hp 菌株稀释为1×109CFU·mL-1悬液灌胃,每只0.5 mL,每2 天1 次,共5 次,灌胃前禁食禁水12 h,灌胃结束后禁食禁水4 h,对照组小鼠以等量生理盐水灌胃;末次灌胃2 周后随机取3 只模型组小鼠和1 只对照组小鼠进断颈处死,取胃组织分为2 份,1 份行常规HE 染色,病理检查分析,1 份行组织匀浆,进行系列稀释后培养,观察Hp定植情况,定植1×105CFU·mL-1及以上判定为定植成功。如2 周不能定植,则灌胃2 周后同样方法观察胃黏膜定植和炎症情况。定植成功则判定为模型建立成功。
1.3 样本采集最后确认感染组有9 只小鼠建模成功。9 只造模成功的小鼠分为S1、S2 和S3 组(每组3 只);将S1、S2 和S3 组小鼠的十二指肠、空肠和结肠命名为SS1、SS2、SS3,SK1、SK2、SK3,SJ1、SJ2 和SJ3;对照组小鼠同样的命名为C1、C2 和C3,并将C1、C2 和C3 的十二指肠、空肠和结肠命名为CS1、CS2、CS3,CK1、CK2、CK3,CJ1、CJ2 和CJ3。
1.4 样品的扩增建库与上机信息采集样品的扩增建库和上机信息采集由生工生物工程(上海)股份有限生物公司完成。
1.5 信息分析方法Illumina HiseqTM得到的原始图像数据文件经碱基识别 (base calling)分析转化为原始测序序列,首先根据overlap 关系进行拼接,区分样本后对序列质量进行质控和过滤,然后进行操作单元分类(operational taxonomic unit,OTU)聚类分析和物种分类学分析。基于OTU 聚类分析结果,进行OTU 主成分分析 (principal component analysis,PCA);基于分类学信息,在各个分类水平上进行群落结构的统计分析;在上述分析的基础上,为了解微生物群落的丰度和多样性,对同肠段组间进行多样性分析 (Alpha 多样性),包括ace 指数、chao 指数、shannon 指数和simpson 指数等统计学分析指数和采用STAMP 软件可获得差异有统计学意义物种,组间比较采用Welch’st-test。利用未观测状态重建的群落系统发育研究(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States,PICRUSt)进行基于京都基因和基因组百科全书(Kyoto Encyclopaedia of Genes and Genomes,KEGG)数据库的功能预测。
1.6 统计学分析采用SPSS 21.0 统计软件进行统计学分析。Alpha 多样性分析采用t检验,2 组间样本均数比较采用两独立样本t检验,样本间Welch’st-test 分析使用错误发现率(false discovery rate,FDR)评估差异的显著性。以P<0.05 为差异有统计学意义。
2 结 果
2.1 OTU 聚类分析本研究共得617 309 条序列数,基于有效序列数据以超过97%相似性进行聚类,共注释5 327 个OTU,每个样本OTU 统计结果见表1。
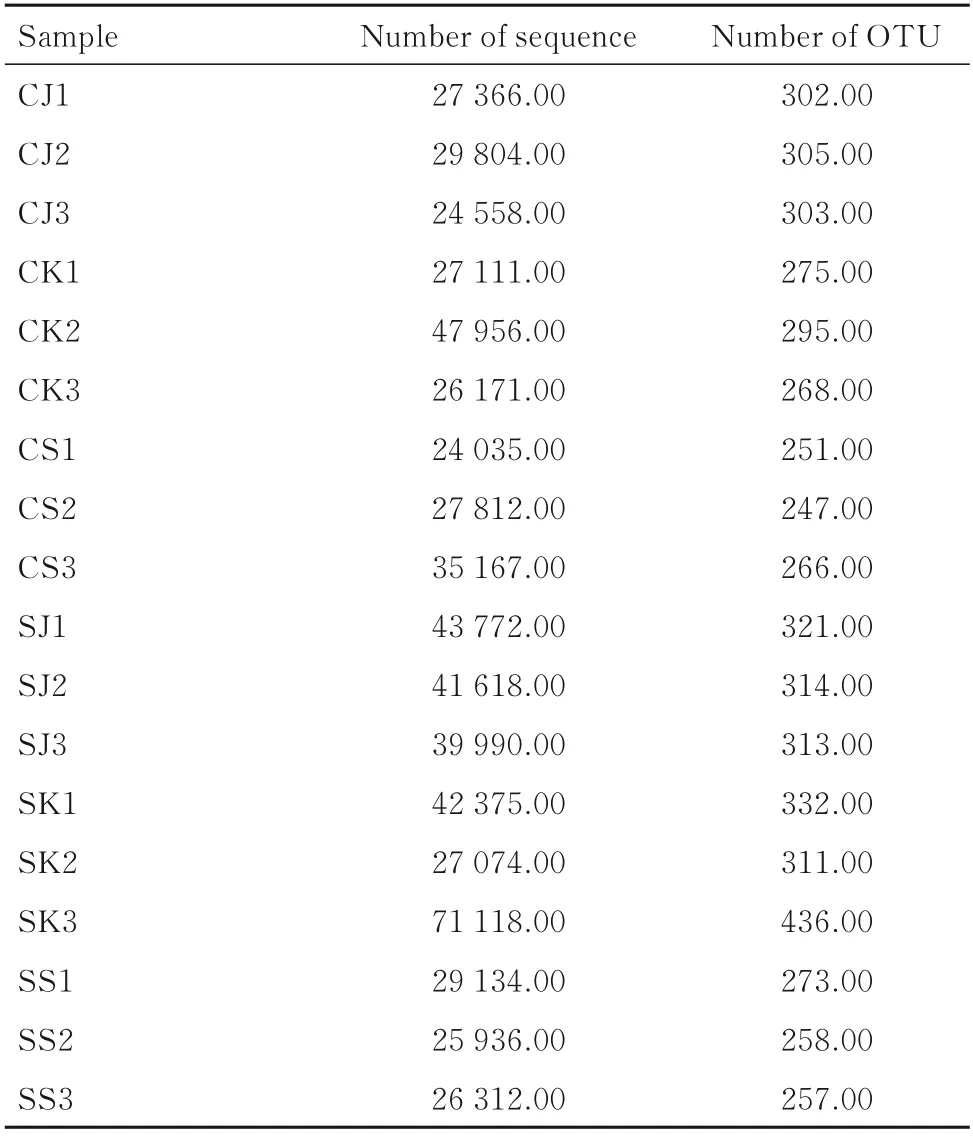
表1 样本 OTU 统计Tab.1 OTU statistics of samples
2.2 2 组样本间物种分布韦恩图以OTU 水平为例,样本间物种分布韦恩图见图1,对照组和感染组小鼠十二指肠、空肠和结肠的OTU 数目为211 个,对照组小鼠十二指肠、空肠和结肠中特有的OTU 数目分别为6、0 和2 个,感染组小鼠十二指肠、空肠和结肠中特有的OTU数目分别为6、4和96 个。

图1 各组样本间OTU 物种分布韦恩图Fig.1 Venn diagrams of species distribution among samples in various groups
2.3 OTU PCA 分析通 过R (v3.6.0)语 言ade4 进行统计分析并作图(图2),横坐标第一主成分最大反映样品间差异的特征值为46.17%;纵坐标第二主成分对最大反映样品间差异的特征值为24.94%。对照组和感染组小鼠肠道各区域大部分分散存在,有部分重合,主成分的离散差距越近,样本物种组成越相似。
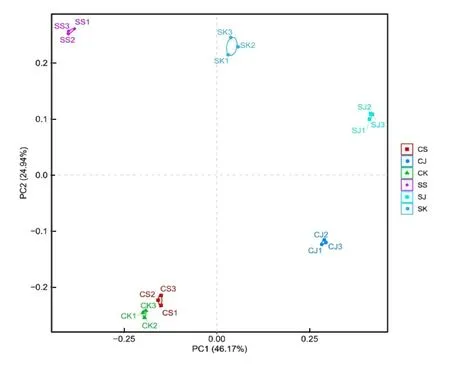
图2 各组OUT 的PCA 分析Fig.2 PCA analysis on OUT in various groups
2.4 微生物种群相对丰度图和优势物种Heatmap图使用R(3.6.0)语言检测样本在不同分类水平上的群落结构,检测出各分类等级的优势菌群丰度和优势物种,见图3 和4。丰度图以门为例见图3A、以属水平为例见图4A,将在所有样本中丰度占比均小于一定比例 (1%)的物种归为其他,其余的作为优势物种进行分析;使用R 语言的gplots package 进行优势物种Heatmap 分析,以门为例见图3B、以属水平为例见图4B。对照组和感染组小鼠十二指肠、空肠和结肠中在门、纲、目、科和属的优势菌均有差异。
在门水平上(图3B),18 个样本共包含7 个门:厚壁菌门、拟杆菌门、放线菌门、变形菌门、疣微菌门、Candidatus-Saccharibacteria菌门和软壁菌门。厚壁菌门、拟杆菌门、放线菌门、变形菌门和疣微菌门在对照组小鼠十二指肠依次占71.14%、22.01%、3.23%、0.85% 和0.01%;在 空 肠 依 次 占 75.90%、16.94%、3.59%、1.77% 和 0.01%;在 结 肠 依 次 占 49.86%、46.08%、2.23%、2.62%和0.29%;在感染组小鼠十二指肠依次占85.04%、10.29%、1.68%、0.85% 和1.11%,在空肠中依次占50.67%、37.30%、3.07%、2.66%和1.90%,在结肠依次占31.47%、60.50%、0.94%、1.71%和0.03%。
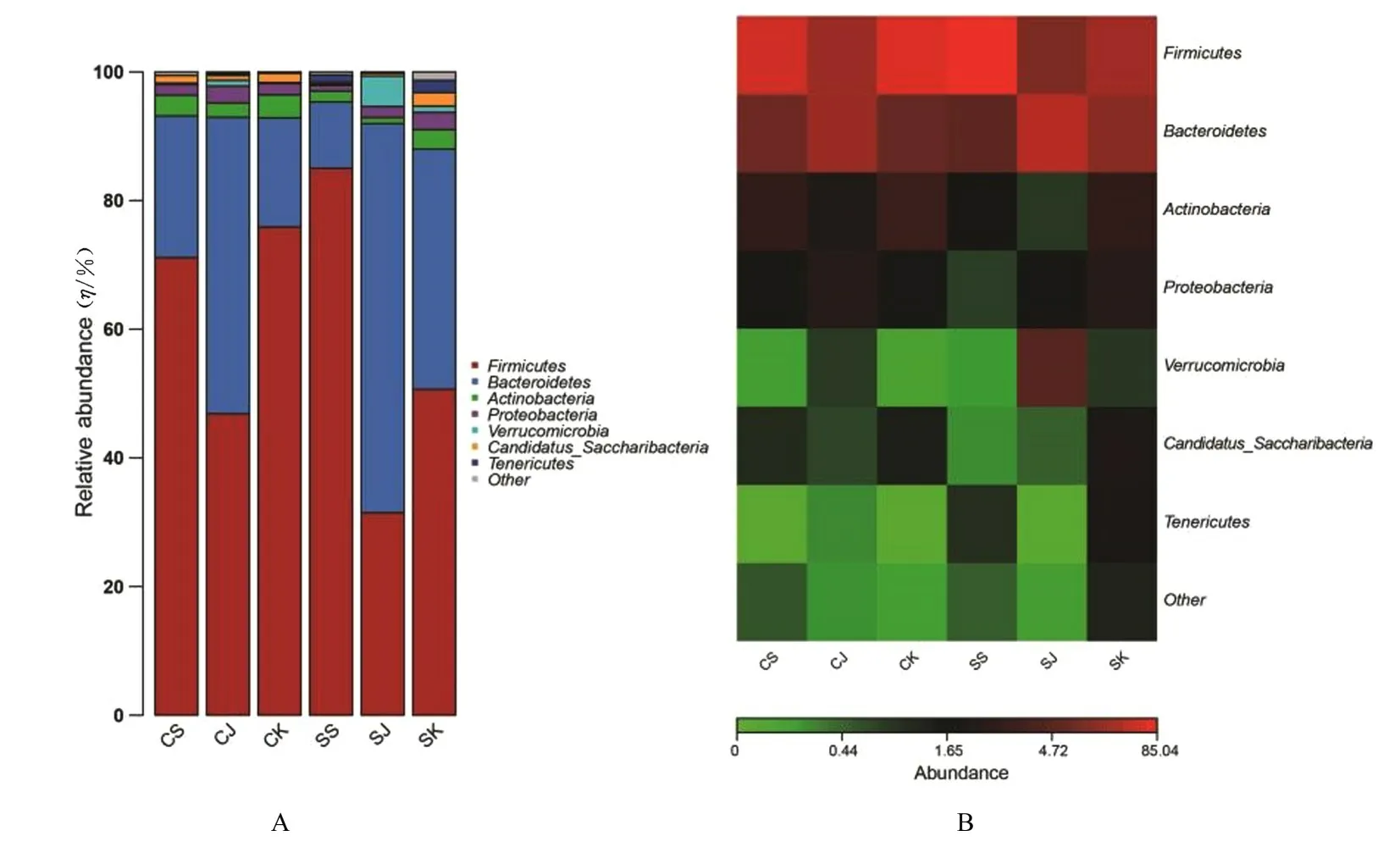
图3 门水平上的微生物种群分布柱状图(A)和丰度热图(B)Fig.3 Histogram of distribution (A)and heat map of abundance (B)of microbial population at phylum level
上述优势菌中的放线菌门多数为益生菌,与对照组比较,感染组小鼠肠道十二指肠、空肠和结肠的放线菌门含量分别低1.55%、0.52%和1.29%。在属水平上(图4B),18 个样本包含21 种优势菌属:乳酸菌属、葡萄球菌属、Clostridium_ⅩⅣa菌属、Turicibacter菌属、阿克曼菌属、双歧杆菌属、Saccharibacteria_genera_incertae_sedis、巴 恩 斯 氏菌属、脱硫弧菌属、另枝菌属和支原体等。

图4 属水平上的微生物种群分布柱状图(A)和丰度热图(B)Fig.4 Histogram of distribution (A)and heat map of abundance (B)of microbial population at genus level
2 组小鼠十二指肠、空肠和结肠中有11 种优势菌属,其在各组的丰度比率见表2。上述优势菌中乳酸菌属和双歧杆菌属多数菌株为益生菌,与对照组比较,感染组小鼠十二指肠、空肠和结肠中乳酸菌属丰度比率明显降低;十二指肠和结肠中双歧杆菌属的丰度比率不同程度降低。支原体为感染过程相关的主要菌种,对照组小鼠未检测到支原体;感染组小鼠十二指肠、空肠和结肠均有不同丰度比率的支原体。

表2 2 组样本中11 个菌属的丰度比率Tab.2 Ratios of abundances of 11 kinds of samples of bacterial in two groups (η/%)
2.5 Alpha 多样性分析对照组和感染组样品多样性统计分析(t检验)结果见表3。shannon 指数中小鼠十二指肠和空肠段菌群的多样性比较差异有统计学意义(P<0.05),感染组细菌的多样性降低;simpson 指数中小鼠肠道各区域差异菌群的多样性比较差异有统计学意义(P<0.01),对照组和感染组其他指数细菌的多样性比较差异无统计学意义(P>0.05)。
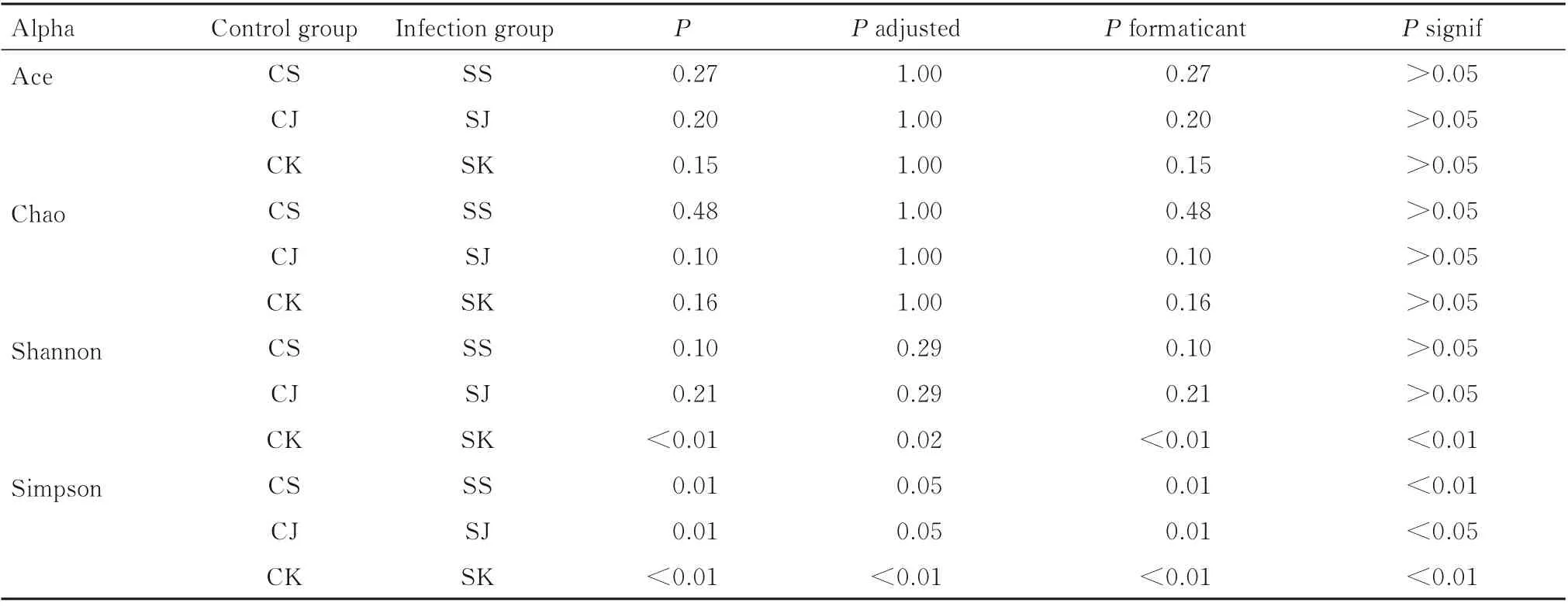
表3 对照组和感染组小鼠肠道菌群Alpha 多样性统计Tab.3 Alpha diversity statistics of intestinal microflora of mice in control group and infection group
2.6 2 组小鼠肠道各区域样本间Welch’st-test 分析分别在各分类等级通过统计学方法检验2 组样本间微生物群落丰度的差异,并使用FDR 评估差异的显著性。对照组与感染组在门水平和属水平上Welch’st-test 差异分析结果分别见图5 和6。图5和6 中间所示为95%置信度区间内物种分类丰度的差异比例,最右边的值为P值,以P<0.05 为差异有统计学意义,用红色标识。此结果与微生物种群相对丰度图和优势物种热图相符合。
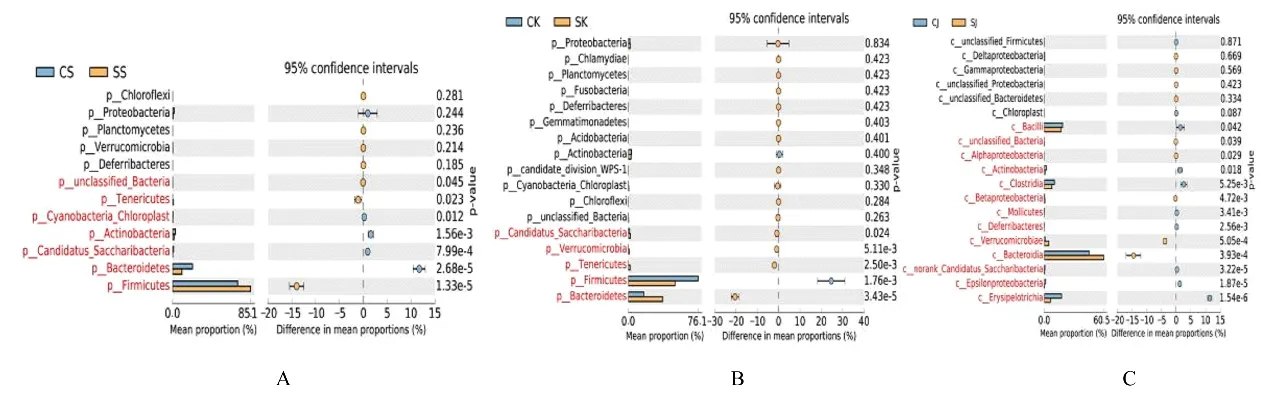
图5 各组小鼠肠道菌群在门水平Welch’st-test 上差异分析结果Fig.5 Difference analysis results of Welch’st-test of intestinal microflora of mice in various groups at phylum level
2.7 功能丰度热图采用 R 语言的gplots package软件,同时利用PICRUSt 算法,基于KEGG 代谢通路数据库对每组样品中的信息绘制功能预测,丰度Heatmap 组间重复性良好,主要富集功能在细菌三磷酸腺苷(adenosine triphosphate,ATP)结合盒B 亚家族、多糖运输系统渗透酶蛋白和能量耦合因子(energy coupling factor,ECF)亚家族RNA聚合酶sigma-70 因子、ABC 结合盒(ATP-binding cassette,ABC)-2 型转运系统ATP 结合蛋白、磷酸甘油酸酯变位酶(EC:5.4.2.1)、抗生素运输系统ATP 结合蛋白、磷酸甘油酸变位酶、多糖运输系统渗透酶蛋白、乳糖操纵子调节蛋白、乳糖阻遏蛋白基因调节家族转录调控因子和多糖运输系统底物结合蛋白等,见图7。

图6 各组小鼠肠道菌群在属水平Welch’st-test 上差异分析结果Fig.6 Difference analysis results of Welch’st-test of intestinal microflora of mice in various groups at genus level
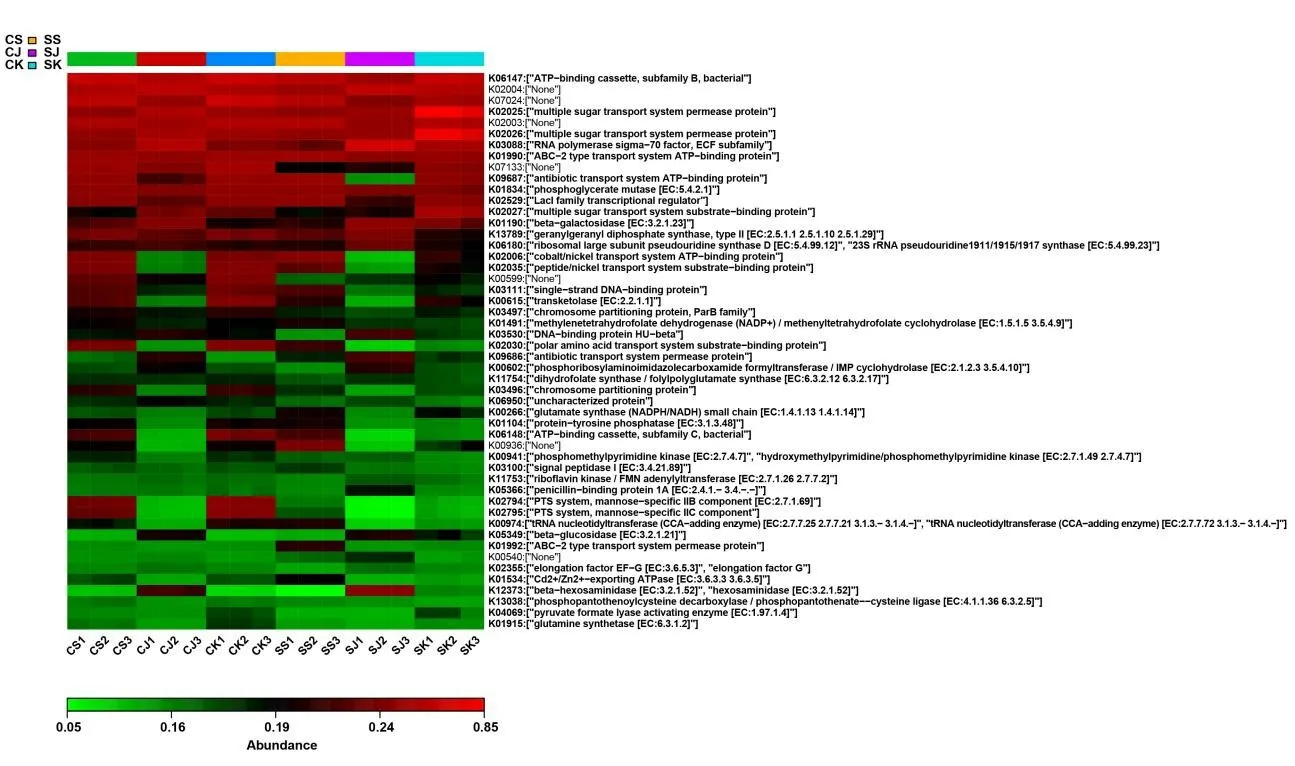
图7 各组功能预测热图(以KEGG 功能注释为例)Fig.7 Heat map of function prediction in various groups(KEGG function annotation as an example)
3 讨 论
胃肠道菌群与其自身宿主相互作用,对健康和疾病发展产生深远影响。放线菌是公认的生物合成工厂[15],其可产生广泛的次级代谢产物,能产生约 70% 的自然抗生素,放线菌有益于宿主的健康[16]。现代高通量测序技术的发展和生物信息学的进步,为研究胃肠道微生物组的多样性和复杂性开辟了一个独立于培养方法的领域,使人类能够深入了解胃肠道微生物[17]。Hp 定植明显影响胃部微环境,进而影响胃部微生物群,并可能与肠道微生物群变化相关,Hp 和肠道菌群之间的相互作用可能在Hp 相关的致癌性和胃外表现中起作用。
本研究对健康和Hp 感染性慢性胃炎C57BL/6小鼠的十二指肠、空肠和结肠中内容物样本进行16SrRNAV3-V4 区信息采集,样本的序列数平均为34 294 条。从物种注释分析和物种热图分布的结果显示:除了未经分类的细菌,与对照组比较,感染组小鼠样本中肠道菌群丰度为空肠>十二指肠>结肠。该变化趋势与美国学者MARTINEZGURYN 等[18]报道的正常肠道菌群丰度为十二指肠>空肠>结肠的结论不一致,可能是Hp 感染导致了上述变化。
为了明确组间菌群种类和丰度差异,本研究在对照组和感染组小鼠肠道各区域菌群中进行OTU PCA 分析、Alpha 多样性参数比较和Welch’st-test分析,与对照组比较,感染组小鼠十二指肠、空肠和结肠放线菌门丰度明显降低,提示Hp 感染后能产生自然抗生素,还能产生各种酶制剂(蛋白酶、淀粉酶和纤维素酶等)、维生素和有机酸,放线菌门生长可能因此受到抑制。与对照组比较,感染组小鼠十二指肠和结肠菌群的乳杆菌属及双歧杆菌含量明显降低,空肠的乳杆菌属含量明显降低,说明感染Hp 后小鼠肠道菌群的乳酸杆菌和双歧杆菌的丰度减少。本研究结果与YIN 等[19]报道的长期感染Hp 蒙古沙鼠十二指肠微环境可能不适合乳酸杆菌繁殖的结论一致。一些研究者认为Hp 感染后乳酸杆菌和双歧杆菌在动物体内丰度减少,是由于慢性Hp 感染小鼠后会诱发胃炎,壁细胞完全萎缩,导致胃内pH 值明显升高(胃酸过少),是胃泌素血症、低氯酸盐血症和“酸性屏障”破坏共同影响所致[20];也有研究者[21]推测可能是神经内分泌途径的影响,如在Hp 感染和诱导免疫病理过程中,在胃中释放的介质(胃泌素、生长抑素、细胞因子和低质子浓度等因素)到肠道中发挥作用,影响了肠道菌群的多样性和丰度。Hp 感染和胃酸过少的患者偶尔会出现非特异性的肠道症状,如不规则的排便、胀气或其他不明原因的腹痛[22-23],可能是由于菌群失调而出现的症状。
通过PICRUSt 功能预测,KEGG 二级代谢通路分析表明:初级和次级代谢物质的运输、转录、坏死性凋亡、免疫应答、炎症反应、氧化还原反应、脂质的相关代谢和糖代谢等的基因通路丰度比较高。本课题组下一步需要对上述主要功能进行相关研究验证。
综上所述,对照组和Hp 感染性慢性胃炎小鼠十二指肠、空肠和结肠的肠道菌群优势菌均存在差异,其中放线菌门、乳酸杆菌和双歧杆菌丰度明显降低,支原体丰度明显升高,本研究结果为今后基于微生物组干预Hp 感染治疗和根除Hp 提供了新的方向。
