肝脏疾病中内质网应激与铁死亡的关系
2023-04-29叶露李秀芹王建青
叶露 李秀芹 王建青
摘要:肝脏疾病的发病机制研究一直广受关注。内质网应激(ERS)是细胞的一种自我保护机制,但持续、严重的应激可诱导凋亡、自噬和铁死亡。其中,铁死亡作为近年研究热点,主要特征为铁依赖性脂质过氧化物的积累,在肝脏疾病的发生发展中发挥着关键作用,但目前关于肝脏疾病中ERS参与铁死亡的相关研究尚少。本文归纳了ERS相关信号通路和铁死亡的发生机制及其在肝脏疾病中的研究进展,为肝病治疗研究提供更多方向。
关键词:肝疾病; 内质网应激; 铁死亡
基金项目:国家自然科学基金(82073566); 安徽省高校优秀青年人才支持计划资助项目 (gxyq2019014); 临床药学与药理学共建项目(2020)
Association between endoplasmic reticulum stress and ferroptosis in liver diseases
YE Lu1,2,3, LI Xiuqin1,2, WANG Jianqing1,2. (1. Department of Pharmacy, The First Affiliated Hospital of Anhui Medical University, Hefei 230012, China; 2. Anhui Public Health Clinical Center, Hefei 230012, China; 3. School of Pharmacy, Anhui Medical University, Hefei 230032, China)
Corresponding author:WANG Jianqing, jianqingwang81@126.com (ORCID:0000-0002-7935-9520)
Abstract:
Research on the pathogenesis of liver diseases has attracted great attention. Endoplasmic reticulum stress (ERS) is a self-protective mechanism of cells, but sustained and severe ERS can induce apoptosis, autophagy, and ferroptosis, among which ferroptosis has been a research hotspot in recent years. Ferroptosis is mainly characterized by the accumulation of iron-dependent lipid peroxides and plays a key role in the development and progression of liver diseases, but there are currently few studies on the involvement of ERS in ferroptosis in liver diseases. This article summarizes the research advances in ERS-related signaling pathways, the mechanism of ferroptosis, and the involvement of ERS in liver diseases, so as to provide more ideas for research on the treatment of liver diseases.
Key words:
Liver Diseases; Endoplasmic Reticulum Stress; Ferroptosis
Research funding:
National Natural Science Foundation of China (82073566); The Program of Excellent Young Talents in Universities of Anhui Province (gxyq2019014); Clinical Pharmacy and Pharmacology (2020)
内质网(endoplasmic reticulum,ER)在维持细胞稳态与机体健康之间的平衡中发挥着重要作用[1]。ER长期处于负荷状态时,其折叠蛋白质的能力下降,进而产生大量未折叠蛋白或错误折叠蛋白,该状态被称为ER应激(endoplasmic reticulum stress,ERS),未折叠蛋白反应(unfolded protein response,UPR)是一种ERS的保护性反应[2]。然而,强烈或持续的ERS仍会影响ER正常的生理功能并导致细胞损伤或死亡[3]。铁死亡是一种区别于凋亡、坏死、自噬的新型细胞死亡方式。其由于谷胱甘肽(glutathione,GSH)过氧化物酶4(glutathione peroxidase 4,GPX4)和GSH等抗氧化系统的功能受损,从而引起脂质过氧化物堆積。最近研究发现,ERS在肝脏疾病的发生、发展过程中往往也伴随着铁超载或脂质过氧化物累积等铁死亡的发生标志。现就ERS通过调控铁死亡影响肝脏疾病的相关研究进展作一综述。
1 ERS的发生机制
当ER出现蛋白折叠错误并积累到临界值以上时即可诱发ERS,并启动UPR来维持ER稳态[4]。UPR可由3种ER跨膜蛋白——肌醇需要酶1α(inositol requiring enzyme 1α,IRE1α)、蛋白激酶样ER激酶(protein kinase RNA-like ER kinase,PERK)和激活转录因子6(activating transcription factor 6,ATF6)调控,当稳态重构失败时ERS将引起细胞死亡[5-6]。
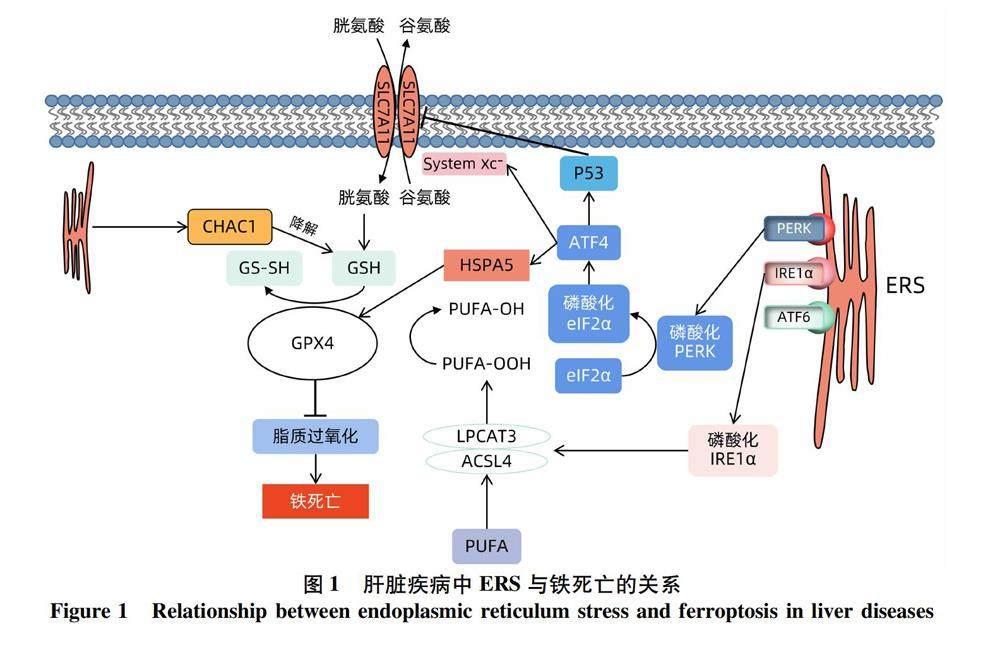
1.1 IRE1α通路 当错误折叠蛋白与IRE1α细胞质尾部的丝氨酸/苏氨酸结构域和核糖核酸酶(ribonuclease,RNase)结构域结合时,IRE1α自磷酸化可导致相邻RNase激活,其RNase将从编码X-box蛋白1(X-box protein 1,XBP1)转录因子的 mRNA 中切除1个 26 nt 内含子以产生稳态转录因子 XBP1s,促进蛋白质折叠能力的恢复、ER相关降解通路的激活、蛋白质分泌基因的转录,进而减轻ER负荷[7-8]。
1.2 PERK通路 PERK同样通过反式磷酸化激活,当其激酶结构域在错误折叠蛋白存在的情况下二聚化时,真核翻译起始因子2α(eukaryotic translation initiation factor 2α,eIF2α)磷酸化。磷酸化后eIF2α 活性被抑制,并因此减慢整体蛋白质翻译,从而使细胞有更多时间处理积压在ER腔内的蛋白质。与此同时,当eIF2α积累到一定程度时将选择性地增加ATF4的表达,其再调控70 kDa热休克蛋白5(recombinant heat shock 70 kDa protein 5,HSPA5)等有利于细胞生存的基因,保护细胞免于死亡[9-10]。
1.3 ATF6通路 ATF6被酶解后,剩余的N端胞质為含有碱性亮氨酸拉链的转录激活功能域,其与XBP1s通过异二聚化的串扰增加靶标的转录,从而扩大ER面积并增加其蛋白质折叠能力以促进细胞存活,并通过调节ER蛋白57等分子伴侣基因的转录,如葡萄糖调节蛋白 78(glucose-regulated protein 78,GRP78)、GRP78/结合免疫球蛋白、C/EBP同源蛋白(C/EBP homologus protein,CHOP),促进错误折叠蛋白的降解[11-12]。
当UPR的3个传感器在尝试重构ER稳态时,其适应性反应不足以恢复其蛋白质折叠能力,则会表现出更强烈的ERS[13-14]。
2 铁死亡的发生机制
铁死亡是一种新型的程序性细胞死亡,该术语于2012年由Stockwell实验室首创,描述了由铁死亡诱导剂(erastin)或铁死亡激活剂(GPX4抑制剂,RSL3)导致的独特类型的细胞死亡[15]。目前,铁死亡发生机制相关研究已颇为全面,其主要发生机制可归纳为以下两点。
2.1 铁代谢异常 正常条件下机体中的铁代谢处于稳态,血液循环中的Fe3+通过转铁蛋白受体进入体内再转化为Fe2+。当发生铁代谢紊乱时,Fe2+在细胞内大量积蓄导致芬顿反应,产生大量脂质过氧化物,进而诱发铁死亡[16]。
2.2 抗氧化系统异常 胱氨酸/谷氨酸逆向转运体(cystine-glutamate antiporter,System xc-)和GPX4目前被认为是介导铁死亡的关键调节因子。胱氨酸可通过System xc-进入细胞内参与GSH的合成,GSH再通过GPX4作用减少活性氧(reactive oxygen species,ROS)的产生,同时GPX4还可将被酰基辅酶A合成酶长链家族成员4(acyl-Co A synthetase longchain family member 4,ACSL4)和溶血磷脂酰胆碱酰基转移酶3(lyso-phosphatidylcholine acyltransferase 3,LPCAT3)氧化成磷脂氢过氧化物的多不饱和脂肪酸(polyunsaturated fatty acid,PUFA)——花生四烯酸、二十二碳四烯酸等还原成无毒的脂质醇。当System xc-或GPX4被抑制时可导致脂质过氧化物积累,是铁死亡的关键信号[17-18]。同时,P53基因作为一种抑癌基因,可通过下调溶质转运家族7A11(solute carrier 7A11,SLC7A11)的表达来抑制System xc-对胱氨酸的摄取,从而影响GPX4的活性,导致抗氧化能力降低,ROS积累和铁死亡[19]。
3 ERS反应促进细胞铁死亡
ERS通过调控相关信号通路可参与自噬、凋亡和铁死亡。自噬作为重要的蛋白质降解途径,可通过降解错误折叠蛋白和清除功能细胞器来缓解ERS。当ERS持续超出临界值且UPR不足以缓解ERS时,UPR过度激活自噬或溶酶体活性异常导致细胞过度降解不能发挥其正常功能而引起细胞凋亡或铁死亡。
Chen等[20]在研究ATF4靶向人脑胶质瘤的实验中发现,ATF4的敲低显著降低了System xc-表达水平,增加了人脑胶质瘤对铁死亡的敏感性。因此,ATF-System xc-通路可能是ERS影响铁死亡的另一条通路。此外,Xu等[21]研究发现,在溃疡性结肠炎患者或葡聚糖硫酸钠诱导的溃疡性结肠炎小鼠模型中观察到铁死亡和ERS反应的发生,当采用PERK的抑制剂GSK414治疗结肠炎小鼠时,葡聚糖硫酸钠引起的铁死亡明显被抑制。Park等[22]在香烟烟雾冷凝物诱导人支气管上皮细胞铁死亡的实验中发现,香烟烟雾冷凝物不仅可以引起铁死亡,同时还激活了ERS中的PERK通路。基因芯片结果分析显示,ERS的发生可增加铁死亡的易感性。
4 ERS参与铁死亡在肝脏相关疾病中的研究现状
目前,铁死亡在肝脏疾病中主要应用于药物性肝损伤、非酒精性脂肪性肝病(NAFLD)和肝细胞癌(HCC),而ERS触发的UPR可通过各种途径参与细胞铁死亡(图1)。因此,对ERS参与铁死亡的发生、发展过程的研究可为肝脏疾病的防治提供有效靶点。
4.1 NAFLD NAFLD是21世纪全球最常见的肝病之一,其发病率仍在逐渐升高[23],但目前其发病机制尚不明确。NAFLD的疾病发展包含从单纯性脂肪变性到非酒精性肝炎(NASH)的一系列疾病,并可进展为肝硬化和HCC,是一种代谢异常综合征的肝损伤,其危险程度不言而喻。
姜嫄等[24]通过GEO数据库的GSE89632数据集和铁死亡数据库(FerrDb)分析NAFLD肝组织中表达差异的铁死亡基因主要在脂肪分化、氧化应激、三价铁(Fe3+)结合方面富集。而在ERS反应的过程中,一些酶和转运蛋白可使进入到细胞质和线粒体基质中的Fe3+转变为Fe2+。Wei等[25]在研究砷诱导成年雄性 Sprague-Dawley大鼠发生NASH时发现,铁死亡关键因子GPX4、GSH下调,ACSL4 mRNA 水平显著上调且细胞线粒体膜破裂和嵴减少或消失,且铁抑素-1治疗砷诱导人肝细胞系L-02细胞时,细胞死亡率明显下降,GPX4的蛋白表达上升,线粒体形态改
善等表明砷诱导NASH中有铁死亡的参与。进一步实验证实,抑制ACSL4是降低砷引发铁死亡的关键。此外,研究也发现大鼠肝脏中磷酸化IRE1α和GRP78的水平上调,提示ERS的发生。采用IRE1α 的强效抑制剂 Irestatin9389预处理细胞后,GPX4 蛋白表达恢复,GSH水平显著上调,丙二醛含量下调,表明砷可通过IRE1α-ACSL4通路参与铁死亡,从而引起NASH。
4.2 HCC HCC是发生在肝细胞或肝内胆管上皮细胞中的恶性肿瘤,其预后较差[26],对HCC发病机制的探索迫在眉睫。
目前,治疗HCC的最有效方式仍是诱导肝癌细胞死亡[27]。Wang等[28]在研究双氢青蒿素(dihydroartemisinin,DHA)治疗肝癌时发现,DHA诱导的4种磷脂酶C(phospholipase,PLC)细胞毒性均可被甲磺酸去铁胺和铁抑素-1治疗,表明DHA可诱导PLC细胞铁死亡。同时PERK、eIF2α、IRE1α等ERS相关因子的表达水平上调,提示UPR信号通路的3个分支均被DHA激活,敲低UPR的3个传感器后,PLC细胞活力增加。DHA通过增加阳离子转运调控样蛋白1抗体(cation transport regulator like protein 1,CHAC1)启动子活性来诱导铁死亡,CHAC1用于降解GSH。在CHAC1上存在ATF4、XBP1和ATF6等多个结合位点,当ATF4(或XBP1、ATF6)表达敲低后,DHA诱导铁死亡的效应显著减弱。综上所述,双氢青蒿素可通过ATF4、XBP1或ATF6诱导的CHAC1表达上调来引发原发性肝癌细胞中的铁死亡事件发生。碳离子辐射对HCC早/中期可提供更好的治疗效果[29]。Zheng等[30]利用碳离子照射联合索拉非尼治疗后,HepG2细胞表现出线粒体萎缩、膜密度增加、波峰降低等铁死亡的形态学特征,同时可见SLC7A11下调,脂质过氧化物产物——丙二醛水平上调,研究表明,碳离子照射可诱导铁死亡。GRP78/结合免疫球蛋白、PERK、ATF4 mRNA水平增加的同时,P53水平也随之上调,表明碳离子照射可诱发ERS,并促进P53基因表达。前期已有研究[31]表明,p53可通过转录抑制SLC7A11促进铁死亡。因此,推测碳离子照射可诱导PERK-ATF4-P53通路来下调SLC7A11促进细胞铁死亡。
4.3 其他肝病 药物性肝损伤是指在药物使用过程中,因药物本身及其代谢产物或由于特殊体质对药物的超敏感性或耐受性降低所导致的肝损伤[32]。其中,对乙酰氨基酚是最主要的致病源,其中毒特征为脂质过氧化物诱导的铁死亡。Tak等[33]在研究肝细胞特异性Gα12蛋白过度表达可能会影响急性肝损伤的实验中发现,ERS可通过IRE1α-XBP1通路反式激活Gα12蛋白,并在后续一系列实验中证实Gα12蛋白是通过诱导花生四烯酸12脂氧合酶 (arachidonate 12-lipoxygenase,ALOX12) 促进脂质过氧化物产生诱发铁死亡,结果表明,ERS中IRE1α-XBP1通路介导Gα12蛋白诱导ALOX12有助于铁死亡。
酒精性肝病是一种大量饮酒导致的肝脏疾病[34]。已有证据[35]发现,在酒精诱导的肝损伤小鼠模型中,肝脏铁过载可诱发ERS。但在ERS的早期阶段,CHOP作为一种新型体内铁调素生产抑制剂,可显著抑制铁调素的表达[36],从而促进铁死亡的发生。因此,酒精性肝病的治疗也可将关注点转向ERS与铁死亡之间的关系。
5 小结
基于ERS参与铁死亡探索肝病治疗方法的模式将受到越来越多的关注。靶向铁死亡在防止由脂质过氧化、炎症浸润和免疫原性介导的各种肝损伤方面发挥重要作用,而ERS的参与也为肝病治疗提供了更多的方法和可能。综上所述,铁死亡是一种在形式和形态上均有别于其他细胞死亡类型的调节性细胞死亡,可受ERS调控。目前,关于ERS参与铁死亡的相关研究仍处于起步阶段,因此,需要进一步明确ERS参与铁死亡对肝脏病理生理学的影响,并积极探索ERS和铁死亡在更多疾病特异性背景下的潜在调节机制。
利益冲突声明:所有作者均声明不存在利益沖突。
作者贡献声明:叶露负责查阅文章,撰写论文;李秀芹、王建青负责修改文章并最后定稿。
参考文献:
[1]WANG M, KAUFMAN RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease[J]. Nature, 2016, 529(7586): 326-335. DOI: 10.1038/nature17041.
[2]HETZ C, PAPA FR. The unfolded protein response and cell fate control[J]. Mol Cell, 2018, 69(2): 169-181. DOI: 10.1016/j.molcel.2017.06.017.
[3]HAN CC, WAN FS. New insights into the role of endoplasmic reticulum stress in breast cancer metastasis[J]. J Breast Cancer, 2018, 21(4): 354-362. DOI: 10.4048/jbc.2018.21.e51.
[4]UDDIN MS, TEWARI D, SHARMA G, et al. Molecular mechanisms of ER stress and UPR in the pathogenesis of alzheimers disease[J]. Mol Neurobiol, 2020, 57(7): 2902-2919. DOI: 10.1007/s12035-020-01929-y.
[5]HETZ C, ZHANG K, KAUFMAN RJ. Mechanisms, regulation and functions of the unfolded protein response[J]. Nat Rev Mol Cell Biol, 2020, 21(8): 421-438. DOI: 10.1038/s41580-020-0250-z.
[6]KOPP MC, LARBURU N, DURAIRAJ V, et al. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor[J]. Nat Struct Mol Biol, 2019, 26(11): 1053-1062. DOI: 10.1038/s41594-019-0324-9.
[7]CALFON M, ZENG H, URANO F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA[J]. Nature, 2002, 415(6867): 92-96. DOI: 10.1038/415092a.
[8]YOSHIDA H, MATSUI T, YAMAMOTO A, et al. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor[J]. Cell, 2001, 107(7): 881-891. DOI: 10.1016/s0092-8674(01)00611-0.
[9]DONNELLY N, GORMAN AM, GUPTA S, et al. The eIF2α kinases: their structures and functions[J]. Cell Mol Life Sci, 2013, 70(19): 3493-3511. DOI: 10.1007/s00018-012-1252-6.
[10]MAMADY H, STOREY KB. Coping with the stress: expression of ATF4, ATF6, and downstream targets in organs of hibernating ground squirrels[J]. Arch Biochem Biophys, 2008, 477(1): 77-85. DOI: 10.1016/j.abb.2008.05.006.
[11]SANTAMARA PG, MAZN MJ, ERASO P, et al. UPR: An upstream signal to EMT induction in cancer[J]. J Clin Med, 2019, 8(5): 624. DOI: 10.3390/jcm8050624.
[12]KIM JM, KIM JS, KIM N, et al. Helicobacter pylori vacuolating cytotoxin induces apoptosis via activation of endoplasmic reticulum stress in dendritic cells[J]. J Gastroenterol Hepatol, 2015, 30(1): 99-108. DOI: 10.1111/jgh.12663.
[13]WALTER P, RON D. The unfolded protein response: from stress pathway to homeostatic regulation[J]. Science, 2011, 334(6059): 1081-1086. DOI: 10.1126/science.1209038.
[14]SHORE GC, PAPA FR, OAKES SA. Signaling cell death from the endoplasmic reticulum stress response[J]. Curr Opin Cell Biol, 2011, 23(2): 143-149. DOI: 10.1016/j.ceb.2010.11.003.
[15]DIXON SJ, LEMBERG KM, LAMPRECHT MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death[J]. Cell, 2012, 149(5): 1060-1072. DOI: 10.1016/j.cell.2012.03.042.
[16]STOCKWELL BR, FRIEDMANN ANGELI JP, BAYIR H, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease[J]. Cell, 2017, 171(2): 273-285. DOI: 10.1016/j.cell.2017.09.021.
[17]KAGAN VE, MAO G, QU F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis[J]. Nat Chem Biol, 2017, 13(1): 81-90. DOI: 10.1038/nchembio.2238.
[18]JIANG L, HICKMAN JH, WANG SJ, et al. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses[J]. Cell Cycle, 2015, 14(18): 2881-2885. DOI: 10.1080/15384101.2015.1068479.
[19]JIANG L, KON N, LI T, et al. Ferroptosis as a p53-mediated activity during tumour suppression[J]. Nature, 2015, 520(7545): 57-62. DOI: 10.1038/nature14344.
[20]CHEN D, FAN Z, RAUH M, et al. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner[J]. Oncogene, 2017, 36(40): 5593-5608. DOI: 10.1038/onc.2017.146.
[21]XU M, TAO J, YANG Y, et al. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis[J]. Cell Death Dis, 2020, 11(2): 86. DOI: 10.1038/s41419-020-2299-1.
[22]PARK EJ, PARK YJ, LEE SJ, et al. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells[J]. Toxicol Lett, 2019, 303: 55-66. DOI: 10.1016/j.toxlet.2018.12.007.
[23]WANG CE, XU WT, GONG J, et al.Progress in the treatment of nonalcoholic fatty liver disease[J]. Clin J Med Offic, 2022, 50(9): 897-899, 903. DOI: 10.16680/j.1671-3826.2022.09.06.
王彩娥, 许文涛, 宫建, 等. 非酒精性脂肪性肝病治疗研究进展[J]. 临床军医杂志, 2022, 50(9): 897-899, 903. DOI: 10.16680/j.1671-3826.2022.09.06.
[24]JIANG Y, HUANG JM, LIANG YZ, et al. Basic study on the mechanism of iron death in non-alcoholic fatty liver disease[J]. J Guangxi Med Univ, 2022, 39(1): 13-20. DOI: 10.16190/j.cnki.45-1211/r.2022.01.003.
姜嫄, 黃锦明, 梁瑜祯, 等. 非酒精性脂肪性肝病铁死亡机制的基础研究[J]. 广西医科大学学报, 2022, 39(1): 13-20. DOI: 10.16190/j.cnki.45-1211/r.2022.01.003.
[25]WEI S, QIU T, WANG N, et al. Ferroptosis mediated by the interaction between Mfn2 and IREα promotes arsenic-induced nonalcoholic steatohepatitis[J]. Environ Res, 2020, 188: 109824. DOI: 10.1016/j.envres.2020.109824.
[26]
WANG KJ, HUANG ZH, SHI QL, et al. Research progress of precise hepatectomy for hepatocellular carcinoma[J]. China Med Herald, 2021, 18(23): 43-46.
王克净, 黄祖鸿, 石清兰, 等. 肝细胞癌精准肝切除的研究进展[J]. 中国医药导报, 2021, 18(23): 43-46.
[27]HASSANNIA B, VANDENABEELE P, VANDEN BERGHE T. Targeting ferroptosis to iron out cancer[J]. Cancer Cell, 2019, 35(6): 830-849. DOI: 10.1016/j.ccell.2019.04.002.
[28]WANG Z, LI M, LIU Y, et al. Dihydroartemisinin triggers ferroptosis in primary liver cancer cells by promoting and unfolded protein response-induced upregulation of CHAC1 expression[J]. Oncol Rep, 2021, 46(5): 240. DOI: 10.3892/or.2021.8191.
[29]SPYCHALSKI P, KOBIELA J, ANTOSZEWSKA M, et al. Patient specific outcomes of charged particle therapy for hepatocellular carcinoma - A systematic review and quantitative analysis[J]. Radiother Oncol, 2019, 132: 127-134. DOI: 10.1016/j.radonc.2018.12.012.
[30]ZHENG X, LIU B, LIU X, et al. PERK regulates the sensitivity of hepatocellular carcinoma cells to high-LET carbon ions via either apoptosis or ferroptosis[J]. J Cancer, 2022, 13(2): 669-680. DOI: 10.7150/jca.61622.
[31]KANG R, KROEMER G, TANG D. The tumor suppressor protein p53 and the ferroptosis network[J]. Free Radic Biol Med, 2019, 133: 162-168. DOI: 10.1016/j.freeradbiomed.2018.05.074.
[32]WANG YW, LIANG YR. Research progress on liver transplantation for drug-induced liver injury[J]. Ogran Transplant, 2022, 13(3): 338-343. DOI: 10.3969/j.issn.1674-7445.2022.03.009.
王砚伟, 梁雨荣. 药物性肝损伤肝移植治疗进展[J]. 器官移植, 2022, 13(3): 338-343. DOI: 10.3969/j.issn.1674-7445.2022.03.009.
[33]TAK J, KIM YS, KIM TH, et al. Gα12 overexpression in hepatocytes by ER stress exacerbates acute liver injury via ROCK1-mediated miR-15a and ALOX12 dysregulation[J]. Theranostics, 2022, 12(4): 1570-1588. DOI: 10.7150/thno.67722.
[34]European Association for the Study of the Liver. EASL clinical practice guidelines: Management of alcohol-related liver disease[J]. J Hepatol, 2018, 69(1): 154-181. DOI: 10.1016/j.jhep.2018.03.018.
[35]CZAJA AJ. Review article: iron disturbances in chronic liver diseases other than haemochromatosis - pathogenic, prognostic, and therapeutic implications[J]. Aliment Pharmacol Ther, 2019, 49(6): 681-701. DOI: 10.1111/apt.15173.
[36]MUELLER K, SUNAMI Y, STUETZLE M, et al. CHOP-mediated hepcidin suppression modulates hepatic iron load[J]. J Pathol, 2013, 231(4): 532-542. DOI: 10.1002/path.4221.
收稿日期:
2022-08-24;錄用日期:2022-10-08
本文编辑:邢翔宇
