高效液相色谱-串联质谱法测定三七中胺苯磺隆残留量的不确定度分析
2023-04-15吴思超李云飞胡湘玲陈庚超
王 英,吴思超,李云飞,吕 平,尤 娜,胡湘玲,程 龙,陈庚超
(1.昆明海关技术中心,昆明 650228;2.黄埔海关技术中心,东莞 523071;3.上海爱博才思分析仪器贸易有限公司,上海 200335)
三七[Panax notoginseng(Burk.)F.H.Chen]是中国传统的名贵中药材,同时也是保健食品,具有活血化瘀[1,2]、抗炎[3]、抗肿瘤[4]、抗血栓[5]、降血脂[6,7]、增强免疫力[8]等功效。为了提高三七的产量,除去三七生长环境周围的杂草,药农通常会使用除草剂胺苯磺隆(Ethametsulfuron)防治杂草危害,导致三七药材中的胺苯磺隆残留对人类健康安全造成威胁。胺苯磺隆,又名油磺隆、菜王星,CAS 号为97780-06-8,分子式为C15H18N6O6S,是由美国杜邦公司20世纪80 年代开发的磺酰脲类长效除草剂,其作用机理是抑制支链氨基酸合成中乙酰乳酸合成酶的活性[9]。胺苯磺隆对多种杂草都具有用量低、防效高、杀草谱广、使用成本低等优点,深受广大用户欢迎[10]。胺苯磺隆检测方法包括超高效液相色谱法-串 联 质 谱(HPLC-MS/MS)法[11]、高 效 液 相 色 谱法[12]、分子印迹技术[13]等。
已有报道鲜见三七中胺苯磺隆残留量不确定度评定,本研究分别依据JJF 1059.1—2012《测量不确定度评定与表示》[14]和JJF 1135—2005《化学分析测量不确定度评定》[15],采用高效液相色谱-串联质谱法对三七中胺苯磺隆残留量的不确定性进行评价,评估各组分的不确定度贡献值,对标准不确定度和扩展不确定度进行计算,为保障检测数据的准确性,促进三七出口贸易提供依据。
1 材料与方法
1.1 样品与试剂
三七,云南鸿翔一心堂药业(集团)股份有限公司;胺苯磺隆(纯度98.5%),坛墨质检科技股份有限公司;甲醇、乙腈(色谱纯),德国Merck 公司;乙酸铵(色谱级),上海阿拉丁生化科技股份有限公司;冰醋酸、氯化钠(分析纯),西陇科学股份有限公司;QuEChERS 方法提取包(无水硫酸镁与无水乙酸钠的混合粉末,4∶1)、QuEChERS 中药农残净化管(无水硫酸镁900 mg、N-丙基乙二胺300 mg、十八烷基硅烷键合硅胶300 mg、石墨化碳黑90 mg),日本岛津公司。
1.2 仪器与设备
4500 型超高效液相色谱-串联质谱联用仪(美国ABSCIEX 公司);XS205DU 型分析天平(瑞士Mettler 公司);Milli-Q 型纯水系统(电导率为18.2 MΏ,美国Millipore 公司);涡旋振荡器(中国Wiggens 公司);3K15 型高速冷冻离心机(德国Sigma 公司)。
1.3 方法
1.3.1 仪器条件
1)色谱方法。Hypersil GOLD C18 色谱柱(100 mm×2.1 nm,1.8 μm);液相流动相流速设为0.3 mL/min;柱温箱温度设为40 ℃;进样体积为10 μL;液相流动相A 为2 mmol/L 乙酸铵水溶液,流动相B 为乙腈;具体流动相洗脱方法见表1。
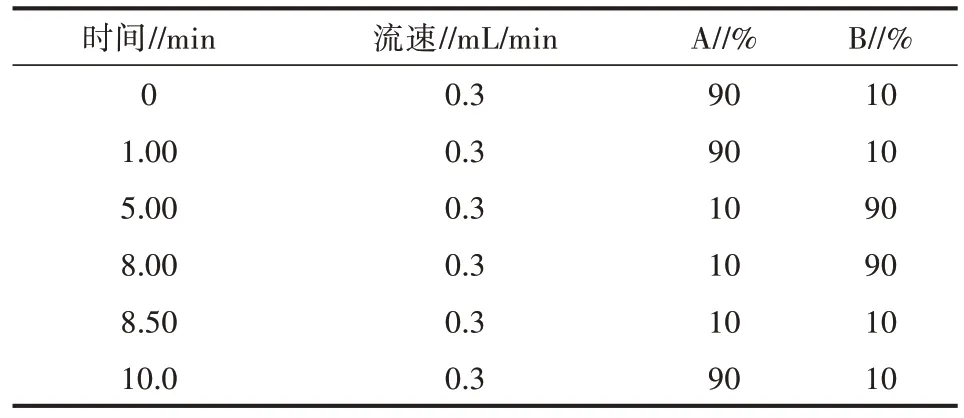
表1 流动相洗脱方法
2)质谱方法。ESI 正离子扫描;多反应监测模式(MRM);驻留时间:50 ms;离子源温度:500 ℃;雾化器压力:35 psi;电喷雾电压:4 500 V;离子对(m/z):294.1/170.1(定性离子)、294.1/137.1(定量离子);锥孔电压:120 V;碰撞电压:24、13 V。
1.3.2 标准溶液的配制 准确称取10 mg(精确至0.01 mg)胺苯磺隆标准品,用乙腈溶解并定容至100 mL,得到质量浓度为0.1 mg/mL 的胺苯磺隆储备液;用乙腈稀释成质量浓度为1 μg/mL 的外标中间液,避光保存于4 ℃冰箱中。
工作溶液:从质量浓度1 μg/mL 的外标中间液中分别准确吸取5、10、25、50、100 μL,用乙腈稀释配制成胺苯磺隆工作液,最终质量浓度分别为5、10、25、50、100 ng/mL。
1.3.3 样品前处理 称取粉碎后过3 号筛(50 目)的三七粉末3 g(精确至0.000 1 g),置于50 mL 离心管中,加入1%冰醋酸溶液15 mL,涡旋振荡1 min,放置30 min,准确加入乙腈15 mL,涡旋混匀,置振荡器上剧烈振荡(500 r/min)10 min,加入QuEChERS 方法提取包,立即摇散,再置于振荡器上剧烈振荡(500 次/min)10 min,离心(4 000 r/min)5 min,取上清液2 mL 放入QuEChERS 中药农残净化管中,涡旋1 min,离心(4 000 r/min)5 min,取上清液过滤,注入高效液相色谱-串联质谱仪,按外标标准曲线法计算。
1.4 数学模型
式中,X为三七中的胺苯磺隆残留量(mg/kg);c为样液中胺苯磺隆的质量浓度(mg/L);v为三七样品的最终定容体积(mL);f为样品前处理稀释倍数;m为三七样品称样量(g)。
2 结果与分析
2.1 不确定度主要来源
通过对样品中胺苯磺隆残留量检测过程进行分析,发现引入不确定度的主要来源包括:①标准品溶液配置;②试样称重;③样品测定过程中,由重复性引入;④高效液相色谱-质谱联用仪引入;⑤标准曲线线性拟合产生;⑥添加回收引入的不确定度[16]。
2.2 各分量不确定度的评定
2.2.1 标准品溶液配制引起的相对不确定度Urel(C)
1)由标准品纯度引起的不确定度[17]。本试验使用的胺苯磺隆标准物质购于坛墨质检科技股份有限公司,其证书显示纯度为98.5%,扩展不确定度为1.2%(k=2),因此,胺苯磺隆标准品的相对标准不确定度为
2)标准物质称量引起的不确定度。使用十万分之一天平精密称取胺苯磺隆对照品10 mg(精确至0.01 mg)。由该分析天平的校准证书可知,该天平的允许误差为±0.5 mg,服从均匀分布,取包含因子
3)标准溶液配制过程中产生的不确定度Urel(C3)。依据JJG 646—2006《移液器》[18]、JJG 196—2006《常用玻璃量器检定规程》[19],不确定度计算采用最大允许误差,按均匀分布,包含因子k= 3,对不确定度开展评价[20],结果见表2。
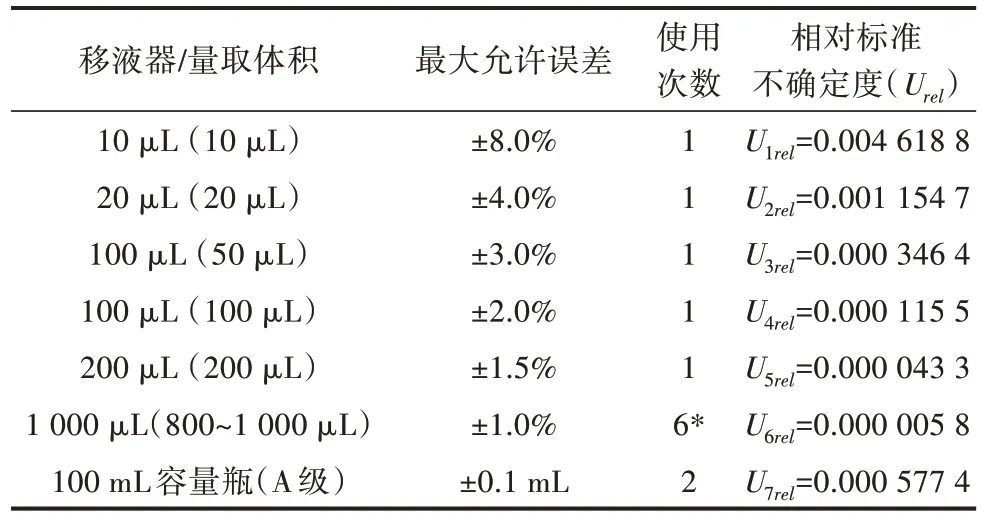
表2 标准溶液配制过程引入的不确定度
在本次标准溶液配制过程中由移液器和容量瓶带来的不确定度合成为[16]:Urel(C3)=0.004 85。
综合以上3 个不确定度分量,标准品溶液配制引入的相对合成不确定度为Urel(C)=
2.2.2 样品称量引入的相对不确定度Urel(m)样品的称样量为3 g 试样(精确至0.01 g)。称量引入的不确定度,按照天平校准证书,天平的最大允许误差计算得到对不确定度为
2.2.3 试样测量重复性引入的相对不确定度Urel(Sˉ)对相同样品6 份,按照相同前处理操作后在相同液相色谱质谱条件下进行检测,样品中胺苯磺隆含量重复性测定结果见表3。
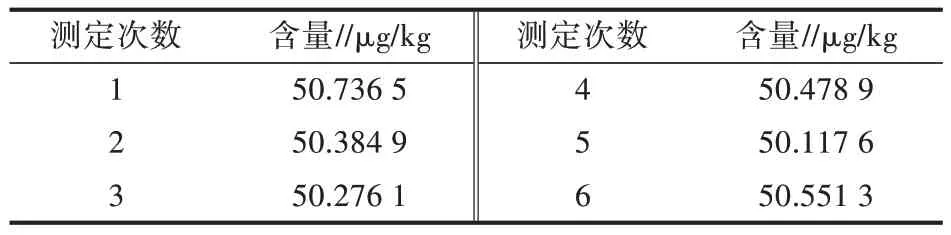
表3 试样测量重复性结果
计算得到标准偏差为0.216 41%,平均含量为50.424 2 μg/kg。因此,重复性测定引起的相对合成不确定度为=0.000 017。
2.2.4 标准曲线产生的相对标准不确定度Urel(Q)对标准溶液浓度与响应值进行拟合[16],计算得到胺苯磺隆回归方程及相关系数,Y=5 147.033 99X+79.368 20,相关系数r=0.995 33,其中截距a=79.368 20,斜率b=5 147.033 99,供试品溶液浓度平均值为50.424 2 μg/kg,计算标准曲线的残差标准偏差SR为:
式(2)中,SR为拟合标准偏差,a为曲线截距,b为曲线斜率,ci为第i次根据标准曲线计算得到的浓度;n为标准溶液的总测量次数,每个标准点测3 次,n=15;yi为第i次测得的色谱峰面积[21]。根据计算得到胺苯磺隆的标准偏差SR=639 260.25。
曲线拟合标准不确定度(UQ)计算公式为[16]:其 中SR为标准曲线的标准差;p为样品平行测定次数,p=6;b为曲线斜率;n=15;c为样品溶液中胺苯磺隆的浓度为胺苯磺隆的平均浓度,ci为第i次根据标准曲线计算得到的胺苯磺隆浓度。因此,
2.2.5 添加回收引入的不确定度Urel(R)取6 份空白三七样品,分别加入0.05 mg/kg 的胺苯磺隆标准溶液,进行加标回收试验[16],试验结果见表4。计算得到标准偏差为3.66%,平均回收率为88.2%,

表4 试样加标回收结果(n=6)
2.3 相对合成标准不确定度
分别将各不确定度分量进行合成[16],得到:Urel0.034 3。样品中胺苯磺隆平均含量为50.424 2 μg/kg,则 其 标 准 不 确 定 度 为U(X)=0.034 3×50.424 2=1.729 6 μg/kg。
2.4 扩展不确定度评定
扩展不确定度由合成标准不确定度乘以包含因子,在95%的置信水平下,取包含因子k=2,则胺苯磺隆测量结果的相对扩展不确定度为U=U(X)×k=1.729 6×2=3.459 2 μg/kg。胺苯磺隆含量X=(50.424 2±3.459 2)μg/kg,k=2。
3 小结
本研究分析了高效液相色谱-串联质谱法测定三七中胺苯磺隆残留量的不确定度,通过计算给出了胺苯磺隆相对合成标准不确定度及扩展不确定度。由评定结果可知,测量结果的不确定度主要由标准溶液配制所引入,添加回收所引入的不确定度次之,其余各成分所引入的不确定度影响较低。因此,应对日常检测工作中的设备仪器定期进行检定校准,严格遵循标准检验规程,以提升检验水平和员工的检测能力,来减小测量不确定度,从而保障检测结果的准确性。
