水稻qPC1基因启动子功能性变异位点的鉴定及其转运功能
2023-03-13何璐璐孔冬艳孙晓宇阿新祥靳可心孙艳芳庞瑞华赵金会汪全秀
彭 波,邱 静,彭 娟,何璐璐,孔冬艳,孙晓宇,刘 岩,阿新祥,靳可心,孙艳芳,庞瑞华,周 伟,赵金会,汪全秀
(1. 信阳师范大学生命科学学院/大别山农业生物资源保护与利用研究院,河南 信阳 464000;2. 信阳市植保植检站,河南 信阳 464000;3. 云南省农业科学院生物技术与种质资源研究所/农业部云南稻种资源观测实验站,昆明 650223)
【研究意义】水稻是世界上最重要的粮食作物之一,全球有超过一半的人口且我国有近2/3的人口以稻米为主食[1-2]。稻米蛋白质是优质蛋白,是人类能量及营养物质的重要来源,改良稻米的营养品质对于改善人类自身的营养与健康具有重要作用。前期,Peng等[3]采用图位克隆策略,已分离克隆到一个正调控水稻种子蛋白质含量的功能基因qPC1(又称OsAAP6基因),且发现qPC1基因的功能性自然遗传变异位点集中在其启动子区的顺式作用元件,但尚不清楚其功能性变异位点及其分子调控机制。进一步进行大田试验,其结果表明在一定范围内,增加田间氮肥浓度,qPC1基因有利于提高水稻籽粒中的氨基酸含量与蛋白质含量,进而改善稻米的营养品质[4]。因此,针对水稻重要品质性状qPC1基因,鉴定其功能性变异位点,开发qPC1分子标记,并探讨其转运功能,为深入揭示qPC1分子调控机制,进而利用qPC1基因开展优质水稻新品种的分子设计育种具有重要的理论意义与潜在的育种价值。【前人研究进展】过去几十年,稻米品质方面的理论研究取得了重要进展,主要围绕其遗传基础与分子调控机理、稻米品质的影响因素与分子改良、基因的分离克隆与功能解析等方面[5-10]。相对于水稻产量不断提高,优质稻米遗传改良的育种实践相对进展缓慢。这是因为稻米品质性状是一个复杂的数量性状,同时受多个基因与环境因素的共同影响[2,11-13]。因此,培育优质水稻新品种是目前国内外育种学家共同追求的重要目标之一。水稻qPC1基因属于氨基酸转运蛋白基因家族中的一员,氨基酸转运蛋白是一类能够介导氨基酸跨膜转运的膜蛋白,在植物生长发育过程中发挥着重要功能[14-16]。目前,针对拟南芥中的转运蛋白研究较多,其中研究最深入的是氨基酸透性酶(Amino acid permease, AAP)家族和类似赖氨酸和组氨酸转运蛋白(Lysine-histidine-like transporters, LHTs)家族,这些家族成员参与了拟南芥体内多种生长发育过程[17-18]。在水稻中,氨基酸转运蛋白基因家族有85个成员,分布于水稻的12条染色体上[19]。其中,氨基酸通透酶参与调控水稻的产量及其品质等多种生物学性状。如OsAAP1、OsAAP3、OsAAP4和OsAAP5与水稻产量性状密切相关[21-23]。qPC1(OsAAP6)可影响水稻中多种氨基酸的分布,是水稻种子蛋白质含量和营养品质的正向调节因子[3-4]。OsAAP7和OsAAP10在水稻中均参与氨基酸的转运,且在OsAAP10突变体中直链淀粉含量与食味值显著下降[24-25]。OsAAP14和OsAAP15参与调控水稻产量,且OsAAP15能够提高极端氮浓度下水稻的产量,可能在水稻育种中具有重要的应用前景[26-27]。水稻OsAAP16在根、叶、花和种子中高表达,并参与转运多种氨基酸[24]。因此,OsAAPs参与了水稻根系中氨基酸的吸收与转运,并参与调节水稻的产量与品质。【本研究切入点】前期,Peng等[3]通过qPC1基因启动子一系列5’端缺失DNA片段与GUS基因融合表达分析,并对qPC1基因5’-UTRs和启动子区域(~1.8 kb)进行比较测序,结合197份来自广泛地理范围的水稻种质资源材料中的种子蛋白质含量测定,结果发现qPC1基因5’-UTR的3个潜在顺式元件所在的两处变异位点(-7~-12 bp、-32 bp)似乎与水稻种子蛋白含量紧密相关。【拟解决的关键问题】本研究采用基因定点突变、水稻原生质体瞬时转化技术和双荧光素酶报告法鉴定qPC1基因启动子功能性变异位点,并利用缺陷型酵母互补试验与水稻根部氨基酸吸收试验解析qPC1的转运功能,以期为水稻新品种的分子设计育种提供新的基因资源。
1 材料与方法
1.1 试验材料
缺陷型酵母互补试验使用酵母菌株24433c和22Δ8AA(中国农业大学李学贤教授课题组惠赠),以及酵母载体pDR195。TA克隆载体pMD18-T和EscherichiacoliDH5α购自Solarbio公司;本研究前期已获得水稻qPC1超量表达阳性[OX(+)]和对照[OX(-)]材料,qPC1抑制表达阳性[RNAi(+)]和对照[RNAi(-)]试验材料,以及珍汕97与南洋占重组自交系群体[3, 28]。其中,珍汕97为籼稻品种(OryzasativaL. ssp.indica),南洋占为粳稻品种(OryzasativaL. ssp.japonica)。
1.2 试验方法
1.2.1 双荧光素酶报告载体的构建 根据载体pGreenII 0800-LUC多克隆位点特征(SmaI-BamH I-SpeI-XbaI-NotI-SacII),结合qPC1基因启动子定点突变序列特征(表1),选择BamH I和NotI两个酶切位点分别加到qPC1启动子5’引物端和3’引物端。利用Primer 5.0软件设计并检测引物(表2)。分别以珍汕97和南洋占幼嫩水稻叶片为材料,采用PCR法扩增目的序列以及定点突变序列,并分别连接到pGreenII 0800-LUC载体。将上述连接产物转化至DH5α中,扩繁并提取质粒pGreenII 0800-LUC-1、pGreenII 0800-LUC-2、pGreenII 0800-LUC-3、pGreenII 0800-LUC-4,即得到最终双荧光素酶报告载体。

表1 qPC1基因定点突变序列

表2 qPC1基因定点突变引物
1.2.2 水稻原生质体的分离及转化 取出20~30棵遮光条件下生长9~12 d的中花11水稻幼苗,切除根部后,将水稻幼茎切成0.5 mm长后迅速置于0.6 mol/L甘露醇中平衡10 min后提取其原生质体。分别吸取10 μL质粒(pGreenII 0800-LUC-1、pGreenII 0800-LUC-2、pGreenII 0800-LUC-3、pGreenII 0800-LUC-4)加至无菌的1.5 mL离心管中,然后缓慢轻柔加入100 μL原生质体悬浮液,轻轻旋转混匀。缓慢沿着离心管壁加入110 μL PEG-CaCl2溶液,将4种质粒分别转入中花11原生质体内,室温避光培养12~16 h。
1.2.3qPC1基因启动子活性检测 根据荧光素酶报告基因检测试剂盒E1910(Premega公司)说明书,将原生质体离心,然后每管加入100 μL 1×PLB后剧烈震荡、破碎细胞。萤火虫荧光测定:12 000 r/min,离心5 min,取10 μL上清液置于预处理过的96孔白色酶标板中,每孔细胞加入LAR II 50 μL,吹打混匀。将96孔白色酶标板放入酶标仪中测定荧光,保存数据并导出。海肾荧光的测定:移出酶标板,向每孔细胞中加入Stop &Glo®Reagent 50 μL,吹打混匀。将96孔白色酶标板放入酶标仪中测定荧光,保存数据并导出。试验设置空白对照,空载体对照,生物学重复3次,技术重复4次。数据处理:每个孔的荧光值(相对活性)=萤火虫荧光值/海肾荧光值。根据检测pGreenII 0800-LUC-1、pGreenII 0800-LUC-2、pGreenII 0800-LUC-3、pGreenII 0800-LUC-4在水稻原生质体内荧光素酶报告基因的表达活性,间接测定对应qPC1启动子活性。
1.2.4 缺陷型酵母表达载体的构建 根据qPC1基因cDNA序列,在qPC15’引物端和3’引物端分别添加限制性内切酶NotI与BamH I序列,采用Bio XM软件设计对应的引物,其引物序列qPC1-F:5’-ATTTGCGGCCGCGAGTATGGACGTGGAGAAGGT GGAGA-3’,qPC1-R: 5’-CGCGGATC CCACCAGCAACTAGTACGAGTACAGCAACA-3’。提取珍汕97的RNA,采用反转录试剂盒(Takara公司,Prime ScriptTMRTMaster Mix)反转录qPC1的cDNA。将其cDNA连接至pMD19-T(Takara公司)载体上得到pMD19-qPC1,然后转化至DH5α(Solarbio,C1100)中,37 ℃过夜培养。提取质粒pMD19-qPC1与酵母pDR195空载体,并进行双酶切(NotI与BamH I),将qPC1基因的cDNA序列连接至pDR195,得到pDR195-qPC1。
1.2.5 水稻根对溶液氨基酸吸收试验 在光照培养箱中,水稻qPC1转基因(超量表达和RNAi抑制表达)种子在生根培养基上无菌生长20 d后,每个家系3株长势一致的小苗进行根吸收试验。吸收溶液是水稻生根培养液但不含氮元素,且用20种氨基酸取代,这些氨基酸的浓度均为50 mmol/L。在吸收试验前对根进行氮饥饿处理2 d,根对氨基酸的吸收速率以6 h内溶液中氨基酸减少的速率来计算,其中溶液中氨基酸含量采用L-8800全自动氨基酸分析仪测定。3次重复,每次溶液中氨基酸含量测定2次。
2 结果与分析
2.1 双荧光素酶报告载体的构建
以水稻品种珍汕97和南阳占DNA为模板,利用定点突变的特异性引物(表1,含有酶切位点BamH I和NotI)进行PCR扩增,分别获得长度为693、440、691、231 bp的qPC1启动子片段,然后分别克隆至pMD18-T载体上。经菌液PCR鉴定得到目的条带大小与预期一致的阳性克隆(图1)。送阳性克隆菌液测序,经测序筛选得到完全无突变的正确序列后,将其分别提取质粒(图2),并克隆至终载体pGreenII 0800-LUC上。经菌液PCR鉴定和测序正确后,对其终载体分别提取质粒并进行琼脂糖凝胶检测和双酶切鉴定(图3),双荧光素酶报告载体pGreenII 0800-LUC-1、pGreenII 0800-LUC-2、pGreenII 0800-LUC-3、pGreenII 0800-LUC-4均构建成功且结果良好。

1、3、4、5泳道同为pMD18-T-1(693 bp);7~12泳道同为pMD18-T-2(440 bp);13~14、16~18泳道同为pMD18-T-3(691 bp);19~24泳道同为pMD18-T-4(231 bp)。The 1, 3, 4 and 5 lanes are the same products of pMD18-T-1 (693 bp); The 7-12 lanes are the same products of pMD18-T-2 (440 bp); The 13-14 and 16-18 lanes are the same products of pMD18-T-3 (691 bp); The 19-24 lanes are the same products of pMD18-T-4 (231 bp).
1~2泳道同为pMD18-T-1(3385 bp)质粒;3~4泳道同为pMD18-T-2(3132 bp)质粒;5~6泳道同为pMD18-T-3(3383 bp)质粒; 7~8泳道同为pMD18-T-4(2923 bp)质粒。The 1-2 lanes are the same plasmids of pMD18-T-1 (3385 bp); The 3-4 lanes are the same plasmids of pMD18-T-2 (3132 bp); The 5-6 lanes are the same plasmids of pMD18-T-3 (3383 bp); The 7-8 lanes are the same plasmids of pMD18-T-2 (2923 bp).
2.2 水稻原生质体转化及启动子活性检测
前期研究结果表明,水稻qPC1基因启动子区有2个共同的自然变异位点(-7~-12 bp,-32 bp),3个潜在顺式作用元件,是引起qPC1基因表达与籽粒蛋白质含量存在差异的主要原因[3]。根据-7~-12 bp,-32 bp两个位点存在的4种突变情况设计定点突变引物(表1,图4),构建双荧光素酶报告基因的载体,瞬时转化水稻中花11原生质体,通过测定荧光素酶活性进而间接测定qPC1启动子活性。
将构建好的双荧光素酶重组载体pGreenII 0800-LUC-1、pGreenII 0800-LUC-2、pGreenII 0800-LUC-3、pGreenII 0800-LUC-4和空载体pGreenII 0800-LUC使用无内毒素质粒提取试剂盒提取高浓度质粒,将双荧光素酶重组质粒和对照空载体分别转化至水稻中花11原生质体中,分别检测其荧光素酶活性。结果发现pGreenII 0800-LUC-1和pGreenII 0800-LUC-2的启动子活性无显著差异(图5),说明当-7 bp处的CACAGA碱基存在时,-32 bp处的碱基突变对转录活性影响不大;pGreenII 0800-LUC-3和pGreenII 0800-LUC-4的启动子活性差异极显著,说明当-7 bp处的CACAGA碱基缺失时,-32 bp处的碱基G突变为C使转录活性显著增加;pGreenII 0800-LUC-1和pGreenII 0800-LUC-4的启动子活性相比,pGreenII 0800-LUC-4的转录活性极显著增加(P<0.001);pGreenII 0800-LUC-2和pGreenII 0800-LUC-3的启动子活性相比,pGreenII 0800-LUC-3的转录活性显著增加(P<0.01)。以上结果表明水稻qPC1启动子转录起始位点上游-7 bp位置处的6 bp(CACAGA)缺失时,qPC1启动子活性显著增加;且当-7 bp处的CACAGA碱基缺失,-32 bp处的碱基G突变为C时,OsAAP6基因启动子活性最强。

M为5000 bp大小的DNA标记;A图中1~2、3~4、5~6泳道分别为pGreenII 0800-LUC-1、pGreenII 0800-LUC-2和pGreenII 0800-LUC-3双酶切产物;B图中1~2为pGreenII 0800-LUC-4双酶切产物。M: DNA marker 5000 bp DNA ladder; A: 1-2、3-4、5-6 are double digested products of pGreenII 0800-LUC-1, pGreenII 0800-LUC-2 and pGreenII 0800-LUC-3, respectively; B: 1-2 are pGreenII 0800-LUC-4 double enzyme cleavage products.

图4 水稻qPC1启动子定点突变位点Fig.4 The site directed mutations in the promoter of qPC1 gene in rice
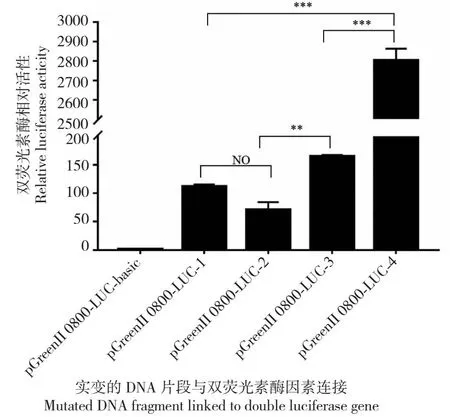
平均值+标准误差;**P<0.01;***P<0.001;NO表示无显著差异。Mean ± sem; **P<0.01; ***P<0.001; NO means no significant difference.
2.3 qPC1功能性分子标记的开发与应用
针对qPC1基因的功能性变异位点,设计对应的分子标记(命名为PB13,表3),并利用分子标记PB13对珍汕97与南洋占的重组自交系群体部分单株进行PCR扩增及测序分析。结果显示,供试单株中有9株来自珍汕97qPC1基因型(231 bp),14株来自南洋占qPC1基因型(237 bp),其余24株为杂合基因型单株(图6)。前期研究结果表明,来源于珍汕97qPC1基因型单株中种子蛋白质显著高于来源于南洋占qPC1基因型的单株[3, 28],且珍汕97qPC1基因序列在其启动子-7~-12 bp位置处,相对于南洋占qPC1基因存在1个6 bp(CACAGA)的缺失。因此,上述试验结果为后期利用qPC1基因进行分子设计育种提供了重要的功能性分子标记。

表3 qPC1基因功能性分子标记
2.4 缺陷型酵母表达载体的构建
以珍汕97的RNA为模版反转录得到其cDNA,利用含有酶切位点(NotI和BamH I)的引物进行PCR扩增,得到预期DNA片段(1543 bp)(图7-A)。将此DNA片段连接至终载体pDR195,经测序和双酶切检测(图7-B),其中泳道1~5表示提取的质粒,大小在7.8 kb左右(与理论大小吻合),泳道6为NotI和BamH I双酶切pDR195-qPC1质粒后的条带,大小分别为6.3和1.5 kb左右(与理论大小吻合),即完成缺陷型酵母表达载体pDR195-qPC1构建。
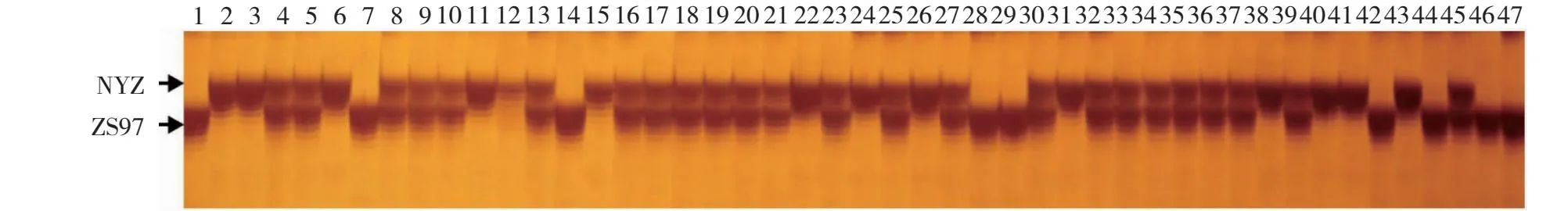
1~47为珍汕97(ZS97)与南洋占(NYZ)的重组自交系群体内的单株,其中1、7、14、18、29、42、44、46和47号样品是珍汕97的基因型;2、3、6、11、12、15、22、24、26、31、38、40、41和43号样品是南洋占的基因型,其余的样品是珍汕97与南洋占杂合基因型。1-47 are individual plants within the recombinant inbred population of Zhenshan 97 (ZS97) and Nanyangzhan (NYZ), among which samples 1, 7, 14, 18, 29, 42, 44, 46 and 47 are genotypes of Zhenshan 97, samples 2, 3, 6, 11, 12, 15, 22, 24, 26, 31, 38, 40, 41 and 43 are genotypes of Nanyangzhan, while the remaining samples are heterozygous genotypes of Zhenshan 97 and Nanyangzhan.
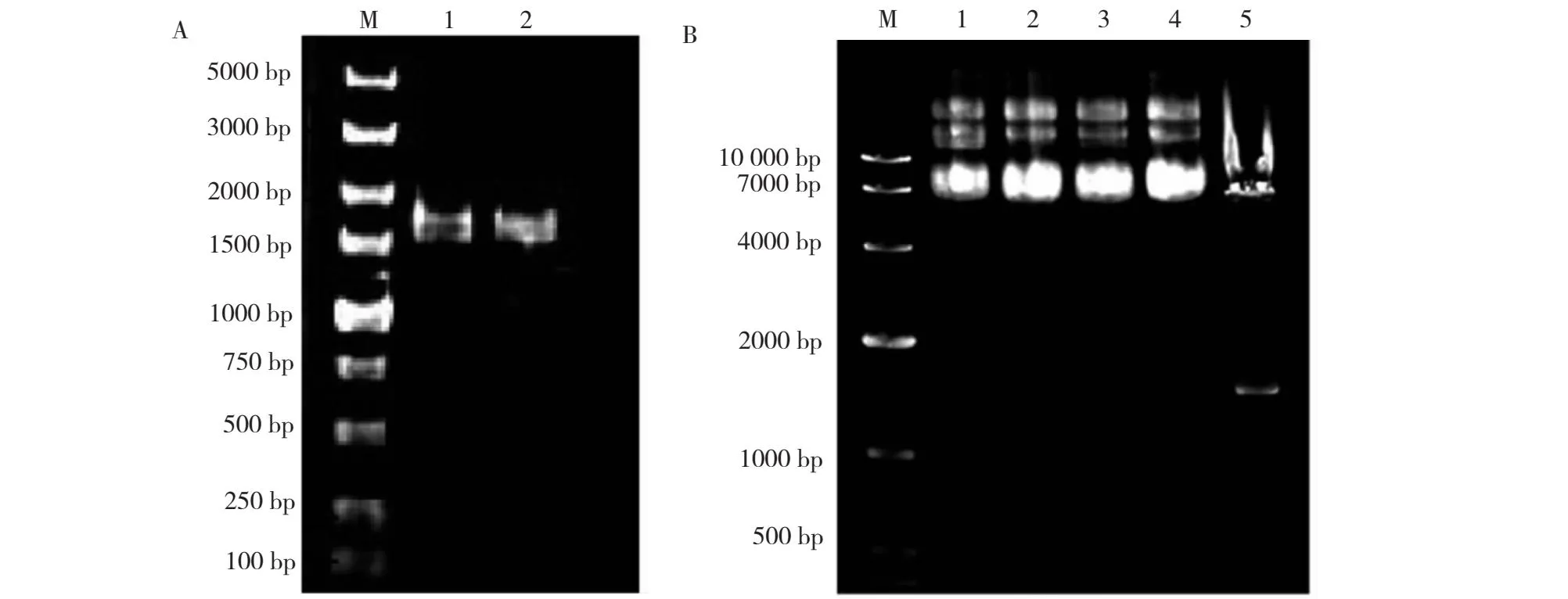
A:1~2泳道为qPC1 cDNA片段;B:1~4泳道为pDR195-qPC1质粒;5泳道为pDR195-qPC1质粒双酶切结果。 A: The 1-2 lanes are the same products of qPC1; B: The 1-4 lanes are the same plasmids of pDR195-qPC1; The line 5 is the double-enzyme digestion of pDR195-qPC1.
2.5 缺陷型酵母生长情况检测
将上述构建好的缺陷型酵母表达载体pDR195-qPC1转入酵母突变体细胞22Δ8AA(qPC1-22Δ8AA),并将pDR195空载体分别转入酵母突变体菌株22Δ8AA(pDR-22Δ8AA)和酵母野生型菌株24433c(pDR-24433c)作为阴性与阳性对照。然后在YNB培养基上培养,结果显示上述3种酵母生长情况良好(图8),即载体均已转入酵母细胞,可以进行后续的氨基酸缺陷型酵母互补试验。
2.6 qPC1在酵母体内转运功能分析
氨基酸缺陷型酵母22Δ8AA对于吸收精氨酸、瓜氨酸、GABA、天冬氨酸、脯氨酸或谷氨酸6种氨基酸存在缺陷,利用酵母22Δ8AA这种特性能够在酵母体内检测qPC1转运功能。将上述构建好的3种酵母(pDR-22Δ8AA、pDR-24433C和qPC1-22Δ8AA)转入酵母完全培养基YPAD中扩繁并筛选。结果显示,qPC1-22Δ8AA酵母生长情况与pDR-24433c酵母阳性对照相似,但明显好于pDR-22Δ8AA阴性对照。在含1 mmol/L谷氨酸和天冬氨酸、以及3 mmol/L精氨酸、脯氨酸、天冬氨酸和L-瓜氨酸的培养基上,qPC1-22Δ8AA酵母生长情况更好,与pDR-22Δ8AA阴性对照存在明显差异。在含3 mmol/L谷氨酸培养基上,pDR-22Δ8AA阴性对照在稀释1000倍时基本不再生长,但qPC1-22Δ8AA酵母在1000和10 000倍时依然生长良好。因此,qPC1-22Δ8AA酵母在供试的6种培养基上均能生长,即qPC1蛋白在体内能够转运多种氨基酸,转运谷氨酸的能力最强(图9)。
2.7 qPC1参与水稻根部氨基酸的吸收与转运
为探究qPC1是否参与水稻根部氨基酸的吸收与转运,将qPC1超量表达(OX)和抑制表达(RNAi)以及对应的对照水稻苗期根部浸入含有20种氨基酸培养液中,根对氨基酸的吸收速率以6 h内溶液中氨基酸减少的速率来计算。结果表明,相对于qPC1超量表达阴性OX (-)植株,脯氨酸、丝氨酸、苏氨酸、甘氨酸、丙氨酸、谷氨酸等酸性氨基酸和总氨基酸含量在qPC1超量阳性OX (+)植株中显著升高,甲硫氨酸则明显下降(图10-A),而在qPC1抑制表达(RNAi)转基因植株中的情况却完全相反(图10-B),即qPC1能够促进水稻根对多种氨基酸的吸收,且对苏氨酸、丝氨酸、甘氨酸、丙氨酸、脯氨酸、谷氨酸和酸性氨基酸具有较快的吸收与转运速率。

A:pDR-24433c;B:pDR-22Δ8AA;C:qPC1-22Δ8AA。
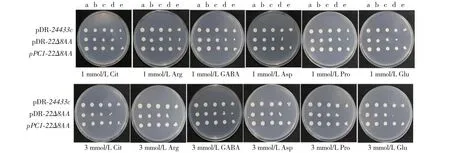
a: OD600值为1.0; b: 稀释10倍;c: 稀释100倍; d: 稀释1000倍; e:稀释10 000倍。a: OD600 value of liquid bacterial germ is 1.0; b: Liquid bacterial germ is diluted 10 times;c: Liquid bacterial germ is diluted 100 times; d: Liquid bacterial germ is diluted 1000 times; e: Liquid bacterial germ is diluted 10 000 times.
3 讨 论
水稻qPC1基因是一个控制稻米蛋白质含量高低的正调控因子,定位于水稻第1染色体上,即qPC1表达水平高时能合成并积累更多的水稻种子储藏蛋白和总的必需氨基酸,最终提高稻米营养品质[3]。大田试验结果表明,随着田间氮肥浓度增加,qPC1表达量逐渐升高,促进稻米中蛋白质、总必需氨基酸和总氨基酸的积累,有利于稻米营养品质的提升[4, 29]。前期研究发现,qPC1基因(又称OsAAP6)在启动子区的变异是引起稻米蛋白质含量变异的主要原因[3]。通过分析197份世界微核心种质资源qPC1基因序列发现,qPC1启动子区存在3个共同的多态性变异位点,且其中2个多态性位点存在3个潜在的顺式作用元件,它们是转录活化因子结合的靶位点[3]。本研究定点突变qPC1启动子翻译起始位点上游-7和-32 bp这2个位点,并将其克隆至双荧光素酶报告基因载体pGreenII 0800-LUC上,将4种重组表达载体瞬时转化至水稻原生质体,检测荧光素酶的活性。结果显示qPC1启动子区-7~-12 bp处6 bp的缺失,能够促进qPC1启动子活性显著增加。因此,qPC1启动子翻译起始位点上游-7和-32 bp处即为qPC1基因的功能性变异位点,并针对该位点设计了对应的分子标记PB13进一步进行了验证,本研究结果为后期全面揭示水稻种子蛋白质含量的分子调控机理提供了重要依据。
氨基酸转运蛋白广泛参与植物生长发育过程,其中拟南芥AtLHT1能够针对组氨酸和赖氨酸进行特异性转运[30],玉米中ZmAAP4可以在体内转运谷氨酸和脯氨酸[31]。作为氨基酸转运蛋白家族成员的一个亚家族,氨基酸通透酶在植物体内能够转运多种氨基酸,参与调控水稻产量与品质。例如,OsAAP1基因通过增加中性氨基酸的摄取和再分布来改善水稻的生长和种子产量[20]。OsAAP4基因参与调节中性氨基酸的分配,能够提高水稻分蘖数及其产量[22]。OsAAP5主要转运碱性氨基酸(赖氨酸和精氨酸)[23]。OsAAP7能够转运甘氨酸、苯丙氨酸、丝氨酸[24]。OsAAP10在水稻中参与中性和酸性氨基酸的转运,并且影响直链淀粉含量与食味值等品质性状[25]。除了天冬氨酸和β-丙氨酸,其余的氨基酸都能被OsAAP16转运[24]。qPC1(OsAAP6)是水稻种子蛋白质含量和营养品质的正向调节因子[3-4],但尚不清楚其转运功能。为探究qPC1的转运功能,将qPC1转入氨基酸缺陷型酵母,结果发现qPC1-22Δ8AA酵母在供试6种培养基上均能生长,即qPC1蛋白能够转运多种氨基酸。为进一步探究qPC1在水稻体内的转运功能,利用qPC1超量表达和抑制表达材料进行水稻根部氨基酸的吸收试验,结果显示qPC1参与水稻根部多种氨基酸的吸收与转运。水稻根部氨基酸的吸收试验和缺陷型酵母的互补试验结果均表明,qPC1蛋白参与多种氨基酸的转运,暗示qPC1蛋白是一种广谱性氨基酸转运蛋白,这为全面解析qPC1基因的生物学功能提供重要线索。

A和B分别表示qPC1超量表达和RNAi抑制表达转基因植株根部氨基酸的吸收试验结果。 (+) 和 (-)分别表示在T2 代转基因阳性和阴性植株;“*”和“**”分别表示在0.05和0.01水平上差异显著。数据以平均值±标准偏差显示,差异基于双尾t-测验,3次生物学重复。插入的表示总氨基酸。A and B represent the absorption results of amino acids in the roots of transgenic plants with qPC1 overexpression and RNAi inhibition, respectively. (+) and (-) represent transgenic positive and negative plants in T2 generation, respectively; ‘*’ and ‘**’ indicate significant differences at the 0.05 and 0.01 levels, respectively. The data is displayed as mean ± standard deviation, with differences based on a two tailed t-test and three biological replicates. The inserted represents the total amino acids.
4 结 论
水稻qPC1启动子区-7~-12 bp位置为其功能性变异位点,设计对应的功能性分子标记PB13并进行验证。qPC1基因参与多种氨基酸的转运,这为后期全面解析qPC1分子调控机制及其利用qPC1基因进行水稻新品种的分子设计育种提供了重要理论依据。
