基于单颗粒电化学碰撞实时监测溶液中纳米金的成核生长过程
2023-02-26白乙艳杨海英秦建芳杨刚张艳清彭得群
白乙艳 杨海英 秦建芳 杨刚 张艳清 彭得群
(运城学院应用化学系,运城 044300)
与宏观物质相比,纳米材料由于表面效应、小尺寸效应、宏观量子隧道效应和量子限域效应而具有独特的光学、电学及磁学等性质。其中,纳米金(Gold nanoparticles,AuNPs)作为最稳定的金属纳米材料之一,已被广泛应用于生物医药[1-3]和能源催化[4-5]等领域。由于成核生长过程会极大地影响纳米材料的晶体结构、尺寸和形貌等性质,而柠檬酸钠还原法是发展最早、应用最广泛的合成AuNPs 的方法之一,所以对其合成机理的研究较多。然而,AuNPs 的成核生长是一个复杂的热力学和动力学过程,不同体系的合成机理也通常不同[6]。目前对于柠檬酸钠还原法合成AuNPs 的机理仍未给出一致的解释,因此,有必要对该体系中AuNPs 成核生长动力学过程的准确监测及合成机理开展深入研究。
传统的原位表征纳米材料的方法,如原位液体透射电镜及紫外-可见吸收光谱等,操作过程中电子束影响、空间受限或系综平均效应等会给测试结果带来一定的误差。随着微纳加工技术和分析方法的发展,高通量检测溶液中的单颗粒成为可能[7]。其中,单颗粒电化学碰撞(Single-nanoparticle electrochemical collision,SNEC)法因具有高分辨、响应快和低成本等优点备受关注[8],其基本原理为:当单个颗粒通过布朗运动扩散到基底电极表面甚至发生碰撞时,会阻碍电极-溶液界面电子传递[9],或本身发生氧化还原反应[10],或催化溶液中的电活性物质发生反应[11],从而产生与单颗粒所对应的电流突变信号,通过分析信号的强度、形状和数目可得到颗粒尺寸、形貌及浓度等信息[12-14]。近年来,SNEC 被用于监测纳米材料的成核生长过程以探究其合成机理[15-20]。Glasscott 等[17]基于SNEC 对金微电极表面电沉积生成的铂原子簇尺寸及催化活性进行了表征,发现通过调控液滴中氯铂酸原液的浓度可有效控制所合成的铂原子簇的大小及催化活性。Bartlett 等[18]将SNEC 与紫外-可见吸收光谱、透射电镜等技术相结合,对由种子银合成棱镜状纳米银的奥斯特-瓦尔德熟化过程进行监测,发现球型种子银在经历圆盘状纳米银中间过程生成棱镜状纳米银后才会急剧生长,带来原子含量上的明显改变。
本研究采用SNEC 法对柠檬酸钠还原法合成AuNPs 的过程进行实时监测,探究其成核生长动力学过程,其中,将氢析出反应(Hydrogen evolution reaction,HER)作为SNEC 实验中的指示反应(见图1)。通过对比不同反应温度下合成的AuNPs,揭示反应温度对其成核生长过程的影响,为可控合成以柠檬酸为配体的AuNPs 提供参考。
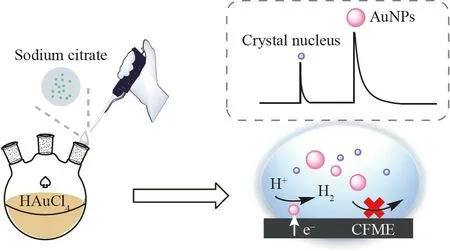
图1 基于单颗粒电化学碰撞(SNEC)监测纳米金(AuNPs)合成过程的示意图Fig.1 Schematic diagram of monitoring the synthesis process of gold nanoparticles (AuNPs) based on single-nanoparticle electrochemical collision (SNEC)
1 实验部分
1.1 仪器与试剂
CHI 660E 电化学工作站和CHI 200B 屏蔽箱及微电流放大器(上海辰华仪器有限公司);Cary5000紫外-可见-近红外分光光度计(美国安捷伦公司);NanoBrook 90Plus Zeta 电位及粒度分析仪(美国布鲁克海文公司);S-4800 扫描电子显微镜(日本日立公司)。
碳纤维(d=7 μm)购自英国顾特服公司;柠檬酸钠和氯金酸(分析纯,上海国药集团化学试剂有限公司);优级纯H2SO4(95%~98%)和HClO4(70%)(洛阳市化学试剂厂);H2O2(30%,天津市大茂化学试剂厂)。实验用水为超纯水(18.2 MΩ∙cm)。
1.2 实验方法
1.2.1 碳纤维微电极(Carbon fiber microelectrode,CFME)的制备
采用火焰封结玻璃密封的方法制备直径7 μm 的CFME。先利用酒精灯加热将硼硅酸盐玻璃毛细管(1.15 mm O.D.,1.0 mm I.D.)拉成两段;然后将直径7 μm 的碳纤维穿入毛细管内,并在酒精灯上烧结,确保碳纤维被毛细管密封;在毛细管内填充石墨后,插入打磨光滑的铜丝,最后利用胶水封结铜丝与毛细管。所制备的CFME 使用前需在水虎鱼溶液中浸泡约20 s,用超纯水冲洗,备用。
1.2.2 AuNPs的制备
采用改良的柠檬酸钠还原法合成AuNPs[21]。室温下,将0.5 mL 1%(m/V)氯金酸加入到50 mL 超纯水中,搅拌下加热至95 ℃,反应5 min 后,在搅拌状态下向其中快速加入0.3 mL 1%(m/V)柠檬酸钠溶液,95 ℃下反应16 min。反应结束后将溶液冷却至室温,然后用截留分子量100 K 的超滤离心管超滤纯化3 次(4000 r/min,4 min)。得到的产物重新分散在2 mmol/L 柠檬酸钠溶液中,4 ℃储存备用。
1.2.3 SNEC测试
将10 mL 0.8 mmol/L HClO4加入到电解池中,然后加入0.2 mL AuNPs 合成过程中所取样品(在AuNPs 合成阶段取样后需立即用冰水冷却,并于4 ℃保存),超声使颗粒在溶液中分散均匀。测试时采用三电极体系,工作电极为CFME,对电极为Pt 丝电极,参比电极为Ag/AgCl(3 mol/L KCl)电极,在–0.6 V 电位下进行i-t测试(采样间隔为3 ms)。碰撞测试时间需控制在30 min 以内以避免AuNPs 团聚对实验结果的影响[14]。
2 结果与讨论
2.1 CFME的制备及表征
本研究采用的工作电极为火焰封结玻璃密封法所制备的圆盘状CFME(图2A 和2B)。如图2C 所示,其在5.0 mmol/L K3Fe(CN)6/0.1 mol/L KCl 溶液中的循环伏安(Cyclic voltammetry,CV)曲线呈“S 型”,这与微电极传质快的特性相吻合[22]。此外,由极限扩散电流可换算得到其有效面积约为38 μm2,满足pmol/L 浓度级的单纳米颗粒检测要求[23]。
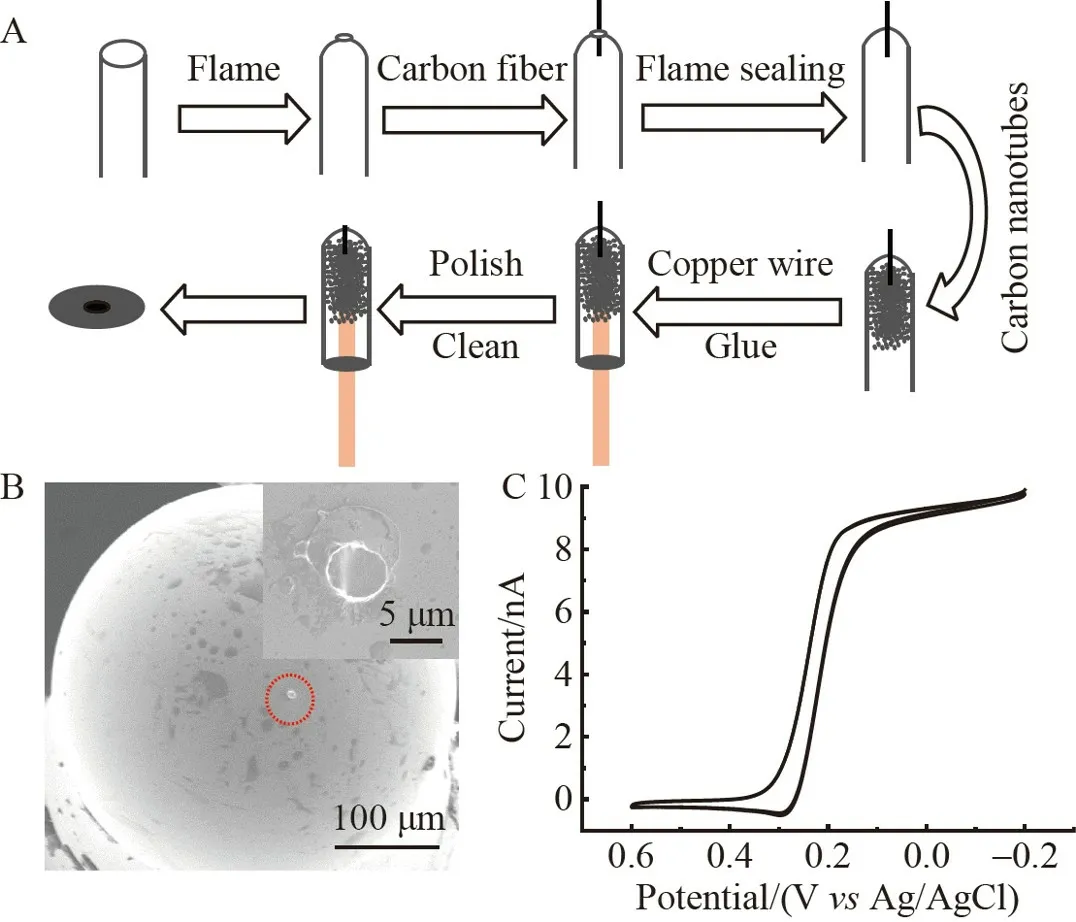
图2 (A)火焰封结玻璃密封法制备碳纤维微电极(CFME)的示意图;(B)CFME 的扫描电子显微镜(SEM)图;(C)CFME 在5.0 mmol/L K3Fe(CN)6/0.1 mol/L KCl 的循环伏安(CV)曲线图,扫速为50 mV/sFig.2 (A)Schematic diagram of preparing carbon fiber microelectrode(CFME)using flame sealed glass sealing method;(B) Scanning electron microscopy (SEM) characterization of CFME;(C) Cyclic voltammogram (CV) of CFME at scan rate of 50 mV/s in 5.0 mmol/L K3Fe(CN)6/0.1 mol/L KCl
2.2 SNEC测试及电压优化
如图3 所示,未加入AuNPs 时,观察不到明显的信号,此时噪声波动约为6 pA;加入约1 pmol/L 50 nm AuNPs 后,出现明显的峰信号,其电流强度不小于噪声波动的3 倍。此结果表明,所观察到的峰信号为AuNPs 碰撞CFME 后催化析氢反应(HER)所产生的电流突变信号。

图3 (A)加入AuNPs 前后碰撞测试i–t 图比较;(B)图3A 中虚线框内的局部放大图Fig.3 (A) Comparison of the i–t curves before and after adding AuNPs;(B) Partially enlarged view within the dashed box in Fig.3A
为确保HER 反应不受电化学反应动力学控制,优化了碰撞测试时施加的电压。如图4A 和4B 所示,当施加电压为–0.4 V 时,出现少量信号峰;当施加的电压负移至–0.6 V 时,信号峰强度变大,数目明显增多。但是,当施加的电压继续负移至–0.8 V 以至–1.0 V 时,信号峰强度几乎不变,信号峰数目却明显减少。由文献[24-25]可知,当纳米颗粒运动到电极表面一定距离(具体取决于双电层厚度的影响)时,纳米颗粒与电极之间的静电作用会明显影响其在电极表面的运动行为。在本研究组的前期工作中同样发现,改变电极电位后颗粒在电极表面运动行为会受到影响,如电极电位由负值变成正值,AuNPs 与电极之间的作用由排斥力变为吸附力,最终导致碰撞信号由峰信号转变为台阶信号[13]。考虑到本研究碰撞测试体系中仅含0.8 mmol/L HClO4,无外加电解质,故存在电迁移影响。因此,如果施加在CFME 上电压太负会导致柠檬酸配体的AuNPs 与CFME 间的静电排斥力增大,从而阻碍其靠近电极。此外,采用火焰封结方法制备了金超微电极(Gold ultramicroelectrode,Au UME,d=30 μm)[13],通过对比CFME 和Au UME 分别作为工作电极时在0.8 mmol/L HClO4溶液中测得的CV 图(图4C)发现,当电压约为–0.6 V 时,Au UME的响应电流约34 nA,而该电位下CFME 电极的响应电流几乎为0,即不发生析氢反应。综上,在本体系中最终选用的施加电压为–0.6 V。
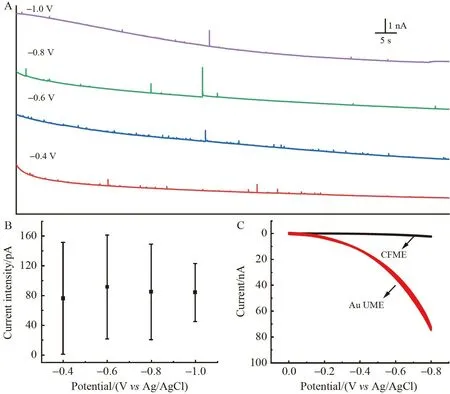
图4 (A)不同施加电压下50 nm AuNPs 在0.8 mmol/L HClO4 中碰撞CFME(d=7 μm)的i-t 曲线;(B)不同施加电压下AuNPs 碰撞信号峰电流强度的比较(n=100);(C)金超微电极(Au UME,d=30 μm)和CFME(d=7 μm)在0.8 mmol/L HClO4 溶液中的CV 图Fig.4 (A) I-t plots of 50 nm AuNPs colliding on CFME (d=7 μm) at various potentials in 0.8 mmol/L HClO4 solution;(B) Comparison of the current intensity under different voltages (n=100);(C) CVs of gold ultramicroelectrode (Au UME, d=30 μm) and CFME (d=7 μm) in 0.8 mmol/L HClO4 solution
2.3 SNEC监测AuNPs合成过程的可行性分析
为探究SNEC 用于监测AuNPs 合成过程的可行性,在加入柠檬酸钠溶液前后分别取样,并在相同条件下进行碰撞测试。如图5A 所示,在加入柠檬酸钠前,即AuNPs 合成体系中只含HAuCl4溶液时,无明显信号峰;加入柠檬酸钠溶液,并在95 ℃下反应16 min 后,出现明显信号峰,表明在柠檬酸钠还原及配位作用下,体系中合成了一定量的球型AuNPs(图5B),其直径约为50 nm(图5C)[26]。在相同条件下对加入柠檬酸钠溶液后不同时间(2、4 和16 min)内的样品进行碰撞测试,并随机统计约300 个信号峰。考虑到纳米颗粒扩散系数与其尺寸成反比,导致通过SNEC 方法更容易检测到小尺寸颗粒,因而对碰撞信号电流强度的频数分布进行加权处理[27]。随着反应时间延长,小信号逐渐减少,大信号逐渐增多,表明合成的AuNPs 尺寸增大(图5D)。上述结果表明,SNEC 可用于监测溶液中AuNPs 的合成过程。

图5 (A)柠檬酸钠溶液加入前后碰撞测试i-t 图;加入柠檬酸钠溶液,并在95 ℃下反应16 min 后溶液的SEM 图(B)和紫外-可见吸收光谱图(C);(D)加入柠檬酸钠溶液加入后,反应2、4 和16 min 后所合成AuNPs 的碰撞信号电流强度加权频数分布图,内插图为加权累计频数分布比较Fig.5 (A) i-t plots before and after adding sodium citrate solution;SEM images (B) and UV-vis absorption spectra(C)of the solution after adding sodium citrate solution and reacting at 95°C for 16 min;(D)Comparison of the weighted frequency distributions of the current intensity for the synthesized AuNPs at 2,4 and 16 min after addition of sodium citrate solution,inset shows weighted cumulative frequency distributions
2.4 基于SNEC实时监测溶液中AuNPs的合成过程
将AuNPs 的合成温度由95 ℃调整为80 ℃,反应不同时间后取样并进行碰撞测试,发现信号峰强度及数目均随反应时间延长而发生改变(图6A 和6B)。如图6C 和6D 所示,各随机统计约300 个信号峰,并对碰撞信号电流强度的频数分布进行加权处理,发现在加入柠檬酸钠溶液0.5 min 后,信号峰电流强度集中分布在18~100 pA 范围内,其中50 pA 以内的信号峰占45%,50~100 pA 范围的信号峰占35%,而近20%的信号峰电流强度分布在100~350 pA 范围内。SEM 表征结果证实此时体系中除了小尺寸晶核外,还包括部分纳米晶体(图7C-a),表明该体系下AuNPs 能快速成核,反应0.5 min 时已成核结束并进入生长初期。

图6 (A)反应不同时间后所合成的AuNPs 的碰撞测试i–t 图;(B)图6A 的局部放大图;反应不同时间后所合成的AuNPs 碰撞信号的电流强度散点图(C)、电流强度加权频数分布图(D)、平均电流强度(E)和碰撞频率(F)Fig.6 (A) Chronoamperometric i–t curves of the synthesized AuNPs with different reaction time;(B) Partially enlarged view of Fig.6A;Scatter plot (C) and weighted frequency distribution (D) of current intensity,average current intensity(E),and collision frequency(F)for the collision signals of the synthesized AuNPs with different reaction time
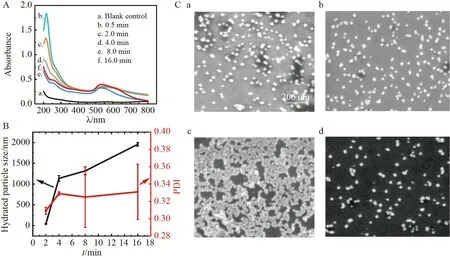
图7 (A)反应不同时间后AuNPs 合成体系的紫外可见吸收光谱;(B)反应不同时间后所合成AuNPs的动态光散射表征;(C)反应0.5(a)、2.0(b)、8.0(c)及16.0 min(d)后所合成AuNPs 的SEM 图Fig.7 (A) UV-vis absorption spectra of the AuNPs synthesis system with different reaction time;(B) Dynamic light scattering characterization of the synthesized AuNPs with different reaction time;(C) SEM images of the synthesized AuNPs after reacting for 0.5 (a),2.0 (b),8.0 (c) and 16.0 min(d)
如图7A 所示,反应2.0 min 后,213 nm 处吸光度明显下降,表明体系中较多Au(Ⅲ)被还原[28]。此时电流强度平均值明显增加(图6E),但100 pA 以内的信号峰却降低至70%(图6D),SEM 表征结果也证实小尺寸晶核变少(图7C-b),碰撞频率几乎不变,这也表明此时体系中颗粒数目未增加(图6F)。因此Au(Ⅲ)并非还原成晶核,而是在晶核表面还原使其快速生长,这与经典的单体扩散(Monomer attachment)生长机理相符。
由图7A 可见,反应4.0 min 后,213 nm 处吸光度明显下降,而50 pA 以内的峰信号却降低至40%(图6D),表明Au(Ⅲ)同样并非被还原成晶核,而是在晶核表面还原使其快速生长。300 pA 以上的峰信号数目及电流强度平均值明显增加(图6D 和6E),表明出现较多大尺寸颗粒,导致测得水合粒径高达1093 nm(图7B)。此外,碰撞频率发生明显变化,由0.35 Hz 下降至0.28 Hz(图6F)。以上结果表明,在2.0~4.0 min 内同时存在AuNPs 之间的聚合生长(Coalescence)[29]。
当反应时间进一步延长至8.0 min 时,213 nm 处吸光度持续下降(图7A),50 pA 以内的峰信号却明显增加(图6D),表明Au(Ⅲ)被还原成晶核。但是,此时碰撞频率几乎不变(图6F),并且开始出现500~800 pA 的信号峰(图6C 和6D),电流强度平均值增大至110 pA(图6E),水合粒径进一步增长至1250 nm(图7B),此时同样存在AuNPs 之间的聚合生长。
如图6D 所示,当加入柠檬酸钠16.0 min 后,50 pA 以内的信号峰数目增长至46%,与0.5 min 时基本相等。SEM 表征结果也证实体系中存在大量小颗粒(图7C-d),但213 nm 处吸光度略微增加(图7A),表明小颗粒的产生不是由于Au(Ⅲ)被还原成晶核。此外,此时开始出现800~950 pA 的信号峰(图6C 和6D),并且水合粒径由1318 nm 增大至1956 nm(图7B),表明8~16 min 内可能发生了奥斯特-瓦尔德熟化(Ostwald ripening),即小颗粒在大颗粒表面溶解,从而大颗粒趋于长大。
此外,通过对比80 ℃与95 ℃时合成AuNPs 的碰撞信号电流强度随反应时间的变化(图6E)可知,80 ℃和95 ℃反应温度下AuNPs 均在16.0 min 内随反应时间延长而逐渐生长变大,并且4.0 min 前的生长速度明显快于4.0 min 后。并且,相同反应时间下80 ℃时的电流强度明显大于95 ℃。这是由于过冷度与临界晶核尺寸成反比,与成核率成正比[30],95 ℃时过冷度较大,因而临界晶核尺寸更小,晶核数目更多,最终导致相同反应时间下合成的AuNPs 尺寸更小。
综上可知,80 ℃下柠檬酸钠还原氯金酸体系中,AuNPs 的合成并非完全按照经典的成核生长理论,而是在不同阶段以不同方式生长,并且通常不止一种生长方式。如图8 所示,首先经历快速成核,并且成核所需时间不超过0.5 min;0.5~2.0 min 内,Au(Ⅲ)在晶核表面还原使其快速生长,与经典的单体扩散生长机理相符;2.0~4.0 min 内,经典的生长与聚合生长方式同时存在,即Au(Ⅲ)在晶核表面还原,此外,多个AuNPs 通过晶面连接在一起;4.0~8.0 min 内,成核与生长同时存在,Au(Ⅲ)被还原成晶核,少部分AuNPs 发生聚合生长;8.0~16.0 min 内,发生了奥斯特-瓦尔德熟化,导致小颗粒与大颗粒数目均增加。
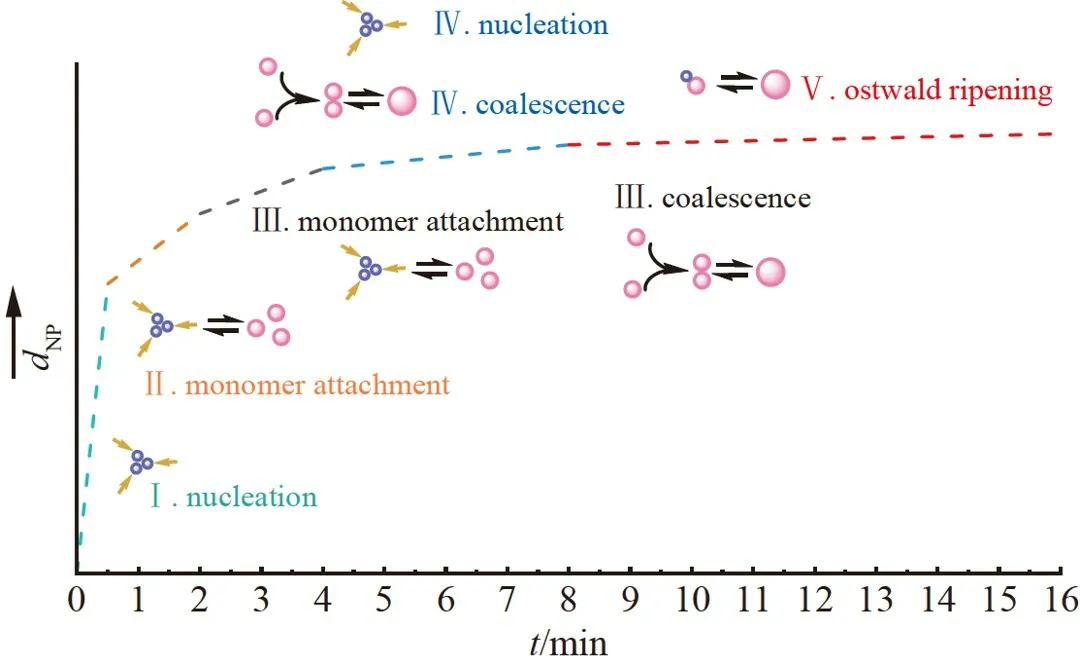
图8 80 ℃下的柠檬酸钠还原氯金酸体系中的AuNPs 合成示意图Fig.8 Schematic illustration for the deduced process of AuNPs formation in the system of sodium citrate reducing chloroauric acid at 80 ℃
3 结论
利用成核生长过程中颗粒尺寸和数目等差异对电流强度及碰撞频率的影响,实时监测溶液中AuNPs的成核生长过程,进而对柠檬酸钠还原法合成AuNPs 的机理进行探究。发现AuNPs 在快速成核后,首先通过经典生长方式即单体扩散,以及聚合生长方式得以快速长大,4.0 min 后通过聚合生长和奥斯特-瓦尔德熟化这两种方式缓慢长大,同时也伴随着成核。此外,与80 ℃相比,相同反应时间下95 ℃时所合成AuNPs 的碰撞电流强度均更小,这是由于此时过冷度较大,临界晶核尺寸更小,晶核数目更多,最终导致相同反应时间下所合成AuNPs 尺寸更小。利用SNEC 监测纳米材料成核生长过程仍处在起步阶段,需进一步探究碰撞信号与颗粒性质之间的关系,进而拓展其在纳米材料合成机理研究中的应用。
