制备液相色谱结合液相色谱-气相色谱联用技术高灵敏检测化妆品中的芳烃矿物油
2023-02-26李婷李冰宁刘玲玲杨梦奇武彦文
李婷 李冰宁 刘玲玲 杨梦奇 武彦文
(北京市科学技术研究院分析测试研究所(北京市理化分析测试中心),北京 100094)
矿物油来源于石油和合成油,涉及C10~C50范围的数以万计不同结构的碳氢化合物,分为烷烃矿物油(Mineral oil saturated hydrocarbons,MOSH)和芳烃矿物油(Mineral oil aromatic hydrocarbons,MOAH)两类[1]。MOSH 和MOAH 的毒性差异很大,其中,MOSH 的生物转化率较低,容易蓄积在动物和人体的脂肪组织、淋巴结、脾脏和肝脏中[1-2]。欧洲食品安全局的风险评估报告指出,MOSH 对人类不构成健康风险[3];而MOAH 特别是三环以上的MOAH 具有潜在的致癌和遗传毒性,应予以高度重视[3-4]。
矿物油是化妆品的重要原料,广泛用于各类护肤品特别是口唇护理产品中,可通过皮肤和口腔进入人体造成危害[5]。2018 年,欧盟要求唇部护理产品中的矿物油必须符合药品要求,使用不含MOAH 的高度精炼矿物油[6]。然而,在市售化妆品中却屡次检出MOAH。2019 年,Zoccali 等[7]检测了意大利生产的9 个化妆品样品,其中4 个样品中检出MOAH,含量高达386~5869 mg/kg;2020 年,Koch 等[8]发现2 个凡士林原料中含有MOAH,其中三环以上的MOAH 含量高达110 mg/kg。
实际上,分析化妆品中的MOAH 非常困难,原因是矿物油中高比例的MOSH 严重干扰MOAH 的分析。目前,矿物油分析方法普遍采用液相色谱-气相色谱联用(LC-GC)技术。然而,LC-GC 的接口限定LC 只能采用250 mm×2.1 mm 的硅胶柱[9],对MOSH 的处理量仅为20 μg,过多的MOSH 将拖尾进入MOAH 馏分,产生假阳性结果[10]。鉴于此,Zoccali 等[7]采用LC-全二维气相色谱-氢火焰离子化检测器/质谱(LC-GC×GC-FID/MS)检测化妆品中的矿物油,将MOAH 的定量限(LOQ)降至11.8 mg/L。然而,该方法过于复杂,难以用于常规检测。Koch 等[8]采用断开LC-GC 接口的策略,将LC 组件单独循环5 轮以制备更多的MOAH 馏分,然后合并、浓缩,注入LC-GC 进行分析,但该方法同样操作繁琐,应用困难。García-Cicourel 等[11]考察了GC-真空紫外检测法(VUV)和超临界流体色谱(SFC)-FID/VUV[12]法,以提高MOAH 分析的灵敏度,但都仅用于化妆品原料分析,未用于市售化妆品的检测。
本研究基于制备液相色谱(Prep-LC)结合LC-GC 建立了化妆品中MOAH 的高灵敏检测方法,通过在样品前处理过程增加Prep-LC 制备环节,降低了分析方法的定量限。本方法可用于化妆品中微量MOAH的日常检测。
1 实验部分
1.1 仪器与试剂
1.1.1 仪器与设备
液相色谱-气相色谱联用仪(LC-GC):LC 20A 液相色谱仪(日本Shimadzu 公司),配备二元泵、紫外检测器(波长230 nm)和Allure Si 色谱柱(250 mm×2.1 mm,5 μm,60 Å,美国Restek 公司);GC 2010 plus 气相色谱仪(日本Shimadzu 公司),配备氢火焰离子化检测器(FID)、预柱(10 m×0.53 mm,美国Restek 公司)和分析柱(15 m×0.25 mm×0.25 μm,美国Restek 公司);LC-GC 接口(德国Axel Semrau 公司)和PAL 自动进样器(瑞士CTC 公司)。
Brave pl700 制备液相色谱仪(Prep-LC,常州磐诺公司):配备二元泵和紫外检测器(波长280 nm),使用Si60 色谱柱(250 mm × 10 mm,5 μm,大连依利特公司)。
GCMS-QP2020 气相色谱-质谱联用仪(日本Shimadzu 公司):配备DB-5HT 石英毛细管柱(15 m×0.25 mm,0.1 μm,美国Agilent 公司)。
A91PLUS 全二维气相色谱-飞行时间质谱仪(GC×GC-TOFMS,常州磐诺公司):一维柱为DB-17HT色谱柱(20 m × 0.25 mm,0.15 μm,美国Agilent 公司),二维柱为DB-1MS 色谱柱(1.5 m × 0.25 mm,0.10 μm,美国Agilent 公司);DV 调制柱(1.3 m×0.25 mm,美国Agilent 公司);SSM1800 调制解调器(上海雪景公司)和EI-TOFMS 0620 飞行时间质谱仪(广州禾信公司)。
Biofuge primo R 低温高速离心机(美国Fisher 公司);SHZ-B 水浴恒温振荡器(上海博迅医疗生物公司);R-100 旋转蒸发仪(瑞士步琦公司);TUS-200P 振荡型恒温金属浴(上海一恒科学公司)。
1.1.2 试剂与标准品
正己烷、二氯甲烷、无水乙醇和甲苯(色谱纯,美国Fisher 公司);无水Na2SO4(分析纯,国药集团化学试剂有限公司)。硅胶(0.063~0.200 mm,德国Merck 公司),使用前在400 ℃下活化16 h,冷却后置于干燥器中,7 d 内使用。
矿物油产品:白油(几乎不含MOAH,美国Sigma-Aldrich 公司);No.45 润滑油(含83.5% MOSH 和9.5%MOAH,中国石油润滑油公司);K009 润滑油(含74.0% MOSH 和11.0% MOAH,德国联邦材料研究和测试研究所)。上述矿物油均用于Prep-LC 的分离性能考察。
标准品:正十一烷(n-C11)和戊基苯(5B),分别用于考察MOSH 和MOAH 是否存在挥发损失;环己基环己烷(Cycy)、2-甲基萘(2-MN)和1,3,5-三叔丁基苯(TBB),MOSH 和MOAH 的定量内标物(可根据实际情况选择2-MN 或TBB);正十三烷(n-C13)和1-甲基萘(1-MN),考察Cycy 和2-MN 是否存在损失或共流出现象;5α-胆甾烷(Cho)、1,4-二(2-乙基己基)苯(DEHB)和苝(Per),用于标记Prep-LC 分离MOSH馏分段的末端、MOAH 的首端和末端;菲(Phe),考察Prep-LC 制备过程是否存在三环以上MOAH 的损失。以上标准品均购自美国Sigma-Aldrich 公司。
配制MOSH/MOAH 混合标准溶液(150~600 μg/mL,溶剂为甲苯,n-C13的浓度为150 μg/mL,n-C11、Cycy、5B、1-MN、2-MN 和TBB 的浓度均为300 μg/mL,DEHB、Phe、Cho 和Per 的浓度均为600 μg/mL),用于LC-GC 分析性能考察和MOAH 测定以及Prep-LC 洗脱条件的优化。
市售的口唇护理与化妆品实际样品共15 个,包括唇膏、口红、唇蜜和凡士林,均购于电商平台。
1.2 实验方法
1.2.1 样品提取
准确称取0.2 g(精确至0.001 g)样品于玻璃离心管中,加入20 μL MOSH/MOAH 混合标准溶液和5 mL 无水乙醇,超声或振荡;加入10 mL 正己烷,以400 r/min 振荡30 min;加入15 mL 去离子水,充分混匀后静置30 min,离心,取上层正己烷相;加入10 mL 正己烷重复提取,合并提取液,减压浓缩至约1 mL,过硅胶柱净化,定容至5 mL,备用。
1.2.2 Prep-LC制备MOAH
进样体积为2.5 mL,流速为3.0 mL/min;以正己烷-二氯甲烷混合溶剂进行梯度洗脱:0~3 min,100%~70%正己烷,保持4 min;7~10 min,由70%正己烷转换为100%二氯甲烷,维持20 min 以再生色谱柱;30~35 min,由100%二氯甲烷转换到100%正己烷,保持20 min 以平衡系统。收集8~15 min 内流出的MOAH 馏分,浓缩至约0.5 mL,备用。
1.2.3 GC-MS分析
进样体积为1 μL,不分流进样;进样口温度为350 ℃;GC 升温程序为:初始温度40 ℃,保持4 min,以20 ℃/min 升至340 ℃,保持10 min;载气为氦气,流速为1.4 mL/min;传输线温度为320 ℃;MS 采用EI 源,以Scan 模式采集,扫描时间为4.0~28 min,采集范围m/z30~700,溶剂延迟4 min。
1.2.4 LC-GC分析
进样体积为50 μL,LC 部分以正己烷-二氯甲烷体系梯度洗脱:0~1.5 min,100%正己烷,1.6~6.2 min,70%正己烷(正向,流速为0.3 mL/min);6.3~15.2 min,100%二氯甲烷(反向,0.5 mL/min);15.3~25.2 min,100%正己烷(正向,0.5 mL/min);25.3~30.0 min,100%正己烷(正向,0.3 mL/min)。其中,2.0~3.5 min 和4.5~6.0 min 分别流出MOSH 和MOAH,经阀(瑞士VIGI 公司)切换,以氢气为载气导入GC 系统。GC 配备两套平行LC-GC 接口,均由预柱、分析柱及其之间通过三通连接的溶剂排空阀(SVE)组成。SVE 在阀切换前0.5 min 开启,转移结束0.3 min 后关闭,期间大部分溶剂通过SVE 排出,浓缩的溶质聚集于GC 分析柱入口,此时启动升温程序:初始温度60 ℃,保持6 min 后,以15 ℃/min 升温至120 ℃,再以25 ℃/min 升温至370 ℃,保持6 min。FID 温度为380 ℃,其辅助气、燃烧气和助燃气分别为氮气、氢气和空气,流速分别为30、40 和400 mL/min。
1.2.5 GC×GC-TOFMS分析
进样体积1 μL,不分流进样;进样口温度为350 ℃;载气为氦气,流速1.7 mL/min;GC 升温程序:初始温度50 ℃,保持3 min 后,以5 ℃/min 升至335 ℃。TOFMS 条件:电子轰击离子源,电离电压–70 eV,离子源温度250 ℃;接口温度320 ℃,检测电压–1640 V;采集质量范围m/z50~650;采集速度为每秒101 张谱;溶剂延迟时间3.0 min。
1.3 数据统计与计算
由于MOAH 的LC-GC 谱图呈鼓包峰,鼓包峰上方的尖峰为干扰物,积分时需扣除,以内标法进行定量分析(内标物可选择2-MN 或TBB)。数据处理分别采用Lab solution、Canvas、Excel 和Origin 软件。
2 结果与讨论
2.1 样品提取
矿物油为弱极性物质,最常用提取溶剂为正己烷[10]。然而,化妆品中含有大量油脂、染料和添加剂等极性物质[13],难溶于正己烷,影响提取效率。本研究先通过乙醇分散并溶解样品,然后加入正己烷,使矿物油完全溶出。乙醇与正己烷互溶,需加水实现相分离,使乙醇-水位于下层,吸取上层正己烷相即得到矿物油提取液[14]。因此,化妆品中矿物油的提取需优化乙醇、正己烷、水的添加量和提取级数。
溶剂的添加量根据样品量确定,样品量由分析方法的定量限(LOQ)决定。目前,国内外还未见化妆品中MOAH 的限量要求。本研究参照文献[8]的研究结果,将LOQ 值预设为10 mg/kg。根据文献[10],进入FID 检测器的矿物油须达到50~100 ng 才能满足分析不确定度要求。因此,以LOQ 值为10 mg/kg 计算得到对应的样品量为0.2 g(见公式(1))。相应地,加入5 mL 乙醇即可分散样品。考察了正己烷的添加量(5、10 和15 mL)对提取率的影响,结果表明,10 和15 mL 正己烷的提取率相当,因此确定正己烷的体积为10 mL。
式中,m为样品质量(g);mi为最少进入FID 的矿物油质量(100 ng);C为预设的LOQ 值,即10 mg/kg;V1为LC-GC 上机试液的定容体积(0.5 mL);V2为Prep-LC 上机试液的定容体积(5.0 mL);Vin1为LC-GC的进样体积(50 μL);Vin2为Prep-LC 的进样体积(2.5 mL)。
为了与后续分析方法匹配并避免浓缩过程造成挥发损失,需要加水除去提取液中的乙醇[14]。实验结果表明,在上述由5 mL 乙醇与10 mL 正己烷组成的混合溶液中,加入15 mL 水可实现分层,吸取上层正己烷相即得到矿物油提取液。为了提取完全,再次加入10 mL 新鲜正己烷进行二次提取。结果表明,第一次提取的MOAH 占两次提取总量的96.6%,即第二次的提取量仅为第一次的3.5%,说明两次提取可以得到几乎全部的MOAH。
2.2 Prep-LC制备MOAH
2.2.1 色谱柱的选择
液相色谱分离的关键是色谱柱,色谱柱填料及其粒径、孔径和柱规格均对色谱柱的分离性能有影响。矿物油的净化普遍采用硅胶。通常情况下,用于固相萃取(Solid phase extraction,SPE)的硅胶存在粒径不均一和分离性能差等问题,无法用于分离MOSH 和MOAH[15];硅胶与银渍硅胶的混合物可以分离MOSH 和MOAH,但稳定性和重现性较差[16]。相比之下,用于LC 的硅胶呈球形,规格均一、刚性较强、分离效能较好,可实现MOSH 和MOAH 的完全分离[10]。
对于液相色谱柱,如果分离填料完全相同,则色谱柱的分离能力与柱规格呈正比关系。如前所述,LC-GC 的硅胶柱规格为250 mm×2.1 mm,仅可分离20 μg MOSH[10],远不能满足化妆品中微量MOAH的检测要求。欧盟法规要求化妆品用矿物油中的MOAH 比例应小于3%(m/m)[17],如果矿物油完全由MOSH 和MOAH 组成,则两者比例不可低于32/1(97/3)。按照预设的LOQ 值(10 mg/kg),0.2 g 样品经过提取、净化和分离,需要注入LC-GC 的MOAH 应为2 μg 以上。因此,需净化去除64 μg(32× 2 μg)以上的MOSH,即Prep-LC 的色谱柱至少需要扩容至LC-GC 硅胶柱的4 倍。因此,常规的250 mm×4.6 mm色谱柱是本方法的最低要求。考虑到欧盟法规仅针对化妆品的原料,而成品的干扰情况更复杂、分析要求更高,因此选择250 mm×10 mm 硅胶柱用于Prep-LC 净化及制备MOAH。
2.2.2 洗脱程序
流动相对色谱柱分离性能的影响很大。在100%正己烷为流动相的情况下,硅胶对极性物质的吸附能力通常最强;加入少量弱极性溶剂,以正己烷-二氯甲烷(7∶3,V/V)混合溶剂洗脱,可实现MOSH 和MOAH 的分离;100%二氯甲烷将极性物质洗脱下来,用于色谱柱的再生[10]。为了绘制洗脱曲线,本研究以MOSH/MOAH 混合标准溶液为研究对象,以正己烷-二氯甲烷(7∶3,V/V)进行梯度洗脱,每3 mL 收集一次馏分并注入GC-MS 进行分析,以筛选Prep-LC 的洗脱条件。
初始条件为:0~8 min,100%正己烷,用于洗脱MOSH;8.1~10 min,由100%正己烷转换为100%二氯甲烷,用于洗脱MOAH;10.1~30 min,100%二氯甲烷,用于色谱柱再生;30.1~35 min,由100%二氯甲烷转换为100%正己烷,并保持到第55 min,用于系统平衡。结果表明,MOSH 馏分于6 min 内全部流出,MOAH 馏分于第9 min 开始流出,即MOSH 和MOAH 流出时间的间隔为2 min,分离效果较好。然而,MOAH 的洗脱时间较长,到第16 min 还未洗脱完全(图1A)。

图1 不同的梯度洗脱条件下MOSH 和MOAH 的流出时间和间隔Fig.1 Elution time and gaps of MOSH and MOAH under different gradient elution conditions
为了缩短MOAH 的流出时间,调整洗脱程序为:0~3 min,100%~30%正己烷,保持7 min,用于洗脱MOSH 和MOAH;10 min 后进入上述再生与平衡程序。结果表明,MOSH 馏分洗脱时间前移了1 min,5 min 内全部流出;MOAH 的开始流出时间也提前至第7 min,到第10 min 全部流出,即MOAH的流出时间为3 min(图1B)。然而,MOSH 和MOAH 的间隔缩短为1 min,较短的间隔时间容易在实际应用中产生交叉干扰。
为了加大间隔时间,调整程序为:0~3 min,由100%正己烷转换为70%正己烷,保持4 min,以洗脱MOSH;7.1 min~10 min,由70%正己烷转换为100%二氯甲烷,保持20 min,用于洗脱MOAH 及再生色谱柱;随后进入上述溶剂转换与平衡程序。结果表明,MOSH 于5 min 内全部流出,MOAH 于第9 min开始流出,到第14 min 结束(图1C),即MOAH 的流出时间为第9~14 min。为了保证回收率,实际操作时前后各增加1 min,即收集第8~15 min 的流出液。此条件下MOSH 和MOAH 间隔3 min,分离效果良好,因此后续实验采用此条件为Prep-LC 的分离条件。
2.2.3 分离性能考察
考察了去除MOSH 的效果与制备MOAH 的回收率。分别以MOSH 和MOAH 混合标准溶液、No.45和K009 润滑油为研究对象,采用1.2.2 节的Prep-LC 分离条件制备MOAH,浓缩后分别注入LC-GC 和GC×GC-TOFMS 进行分析。结果表明,LC-GC 谱图中未出现n-C11、n-C13、Cycy 和Cho 等代表MOSH 馏分的化合物,也未检出润滑油中的MOSH,说明采用Prep-LC 制备的MOAH 馏分较纯净,不存在MOSH 残留。类似地,各样品的MOAH 馏分的GC×GC-TOFMS 谱图也未出现MOSH 的峰(电子版文后支持信息图S1),说明采用Prep-LC 可以有效去除MOSH。
通过比较Prep-LC 制备前后的MOAH 含量计算回收率。结果表明(表1),代表MOAH 馏分的7 种化合物即DEHB、5B、1-MN、2-MN、TBB、Per 和Phe 的回收率为92.7%~104.0%,润滑油No.45 和K009中MOAH 的回收率则均高于98%。其中,5B 的回收率(92.7%)偏低,可能是在浓缩和进样过程发生了挥发损失;Per(4 个苯环)的回收率(92.8%)也偏低,但Phe(3 个苯环)却未受影响,说明Prep-LC 的硅胶柱对高环数稠环芳烃的吸附作用较强,造成洗脱不完全。矿物油中的MOAH 主要由高度烷基化的三环以下芳烃组成,因此Prep-LC 处理对MOAH 影响较小。
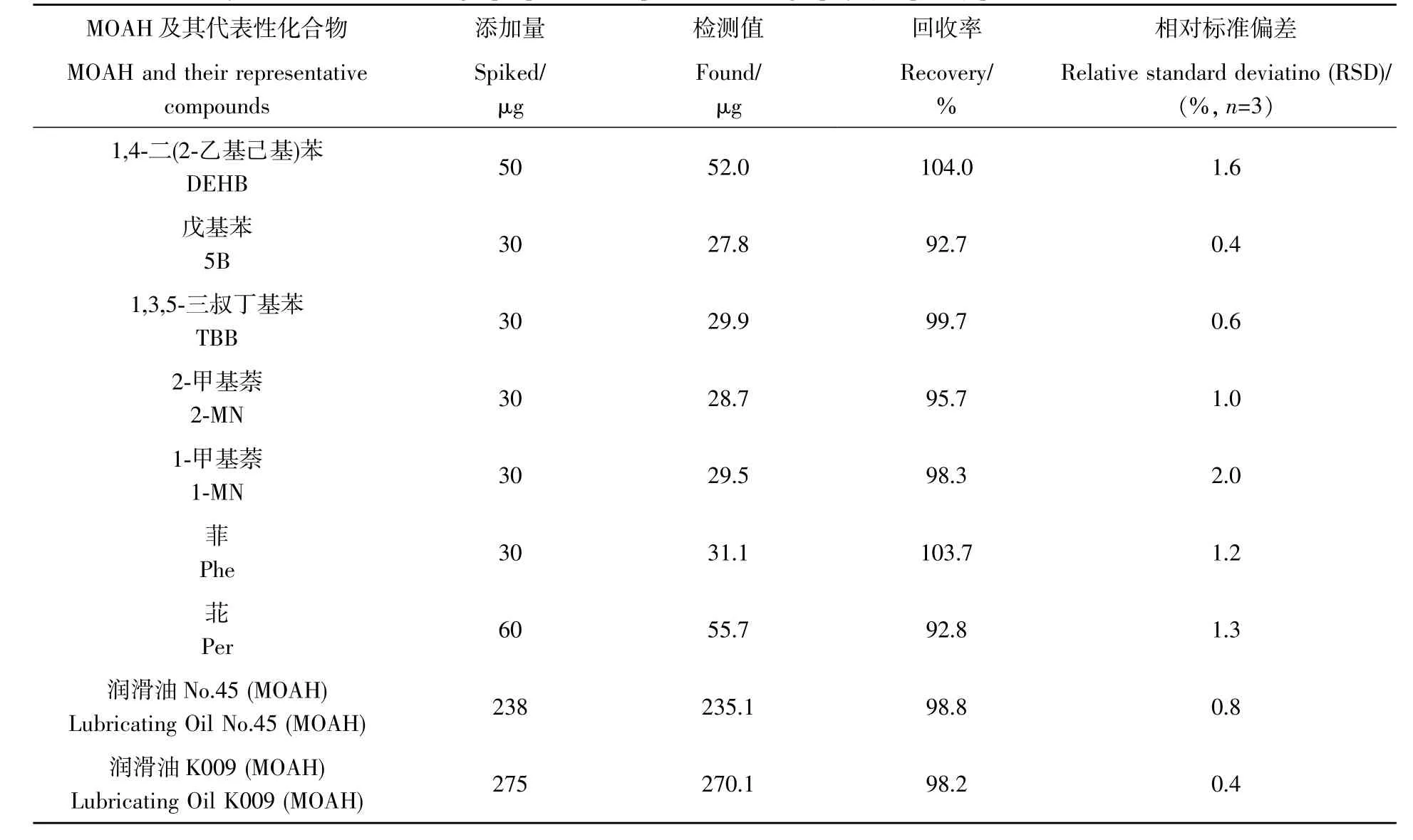
表1 制备液相色谱(Prep-LC)净化MOAH的回收率Table 1 Recovery of the MOAH through preparative liquid chromatography (Prep-LC) purification
2.2.4 净化容量考察
为了考察Prep-LC 对MOSH 的分离容量,以白油为研究对象,逐步增大其进样量,然后收集MOAH馏分并以LC-GC 考察MOSH 残留情况。结果表明,当白油的进样量增至2.4 mg 时,LC-GC 均未检出MOSH;白油的进样量继续加至3.0 mg 时,其MOAH 馏分的LC-GC 谱图中出现MOSH 鼓包峰,说明MOSH 发生拖尾进入MOAH 馏分,因此确定Prep-LC 最多可以去除2.4 mg MOSH。
色谱柱的净化容量与其规格密切相关。LC-GC 的色谱柱为250 mm×2.1 mm,可分离20 μg MOSH;将柱规格增大为250 mm × 10 mm,理论上可净化约450 μg MOSH。然而,本研究中Prep-LC 最多可去除2.4 mg MOSH,是该理论值的5 倍以上,说明增大色谱柱对提升净化容量有叠加效应,类似的结论在文献[18]中也得到证实。但是,为了保证净化效果,实际应用时通常需要设定“安全范围(Safety margin)”,因此建议MOSH 的净化容量不超过2.0 mg。
2.3 方法学考察
本研究采用LC-GC 检测,所用FID 检测器的线性范围较宽,但LC-GC 接口技术限制了LC 的容量,使得进入GC 的矿物油远未达到FID 的上限。因此,矿物油分析的方法学考察一般不涉及线性范围[10]。
由于MOAH 由数以万计不同结构的芳烃化合物组成,其分析谱图呈较宽沸程的鼓包峰形状,并且不同来源MOAH 的沸程范围差别很大。为了满足分析不确定度要求,通常进入FID 的MOAH 应达到50~100 ng[10]。本研究以此为依据,预设定量限为10 mg/kg 并计算出称样量为0.2 g(计算方法见公式(1))。为了考察本方法的实际定量限,按照1.2 节实验方法对空白样品进行了分析。结果表明,LOQ即空白样品响应信号的标准偏差10 倍对应的浓度为4.5 mg/kg,考虑到实际样品的干扰物差异和MOAH谱图的碳数差异,确定本方法的LOQ 值为10 mg/kg。
采用标准添加法考察回收率。由于目前尚无MOAH 的标准品,本研究以K009 润滑油中的MOAH作为标准添加物。在空白样品中添加不同浓度的MOAH,添加浓度分别为LOQ 的1、2、5 倍,即10、20 和50 mg/kg。结果表明,加标回收率为82.0%~100.8%,相对标准偏差(RSD)为3.6%~7.3%(表2)。由于目前国内外还没有化妆品中微量矿物油的分析标准,本研究参考欧洲联合研究中心(Joint Research Center,JRC)[19]关于食品与食品接触材料中矿物油分析的方法指南,其中要求回收率为80%~110%,RSD <10%。因此,本研究建立的方法可以满足实际样品中矿物油分析的要求。
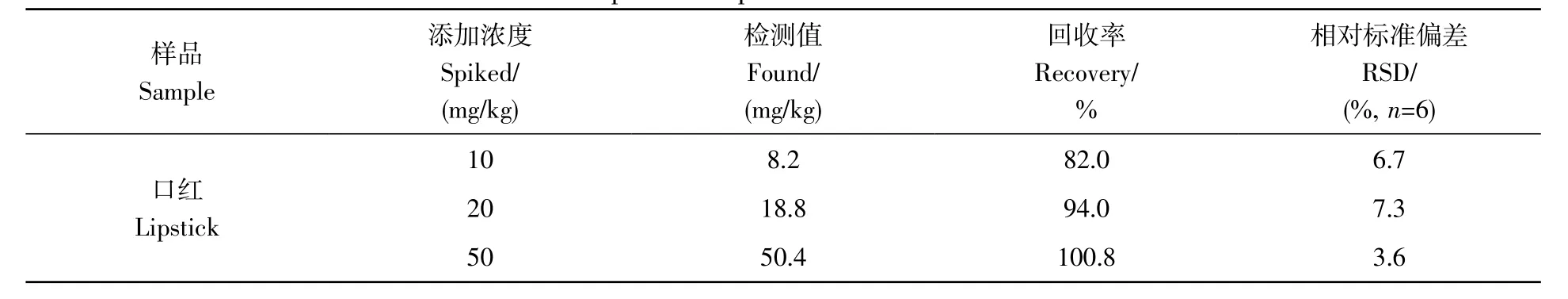
表2 口红样品中MOAH的加标回收率和相对标准偏差Table 2 Recoveries and RSDs of MOAH in a lipstick sample
2.4 实际应用
采用本方法对市售15 个口唇护理与化妆品样品中的MOAH 含量进行了检测,结果表明,其中4 个检出MOAH,含量为204~1460 mg/kg(图2),此值与在意大利和德国的调查结果类似[7-8]。考察了阳性样品对应的MOSH 谱图,发现两类矿物油的碳数范围相同,根据JRC 方法指南,可确定其为矿物油污染物[19]。此外,4 个阳性样本的MOAH 谱图的碳数范围和形状差别较大,说明其来源于不同的矿物油产品。不同于其它3 个样本,图2C 中出现2 个鼓包峰,说明其MOAH 可能来自2 种不同的矿物油。另外,尽管这些阳性样本的碳数范围不同,但其最大碳数均超过C50,说明这些化妆品的污染可能来源于高沸点的润滑油或润滑脂原料[10]。
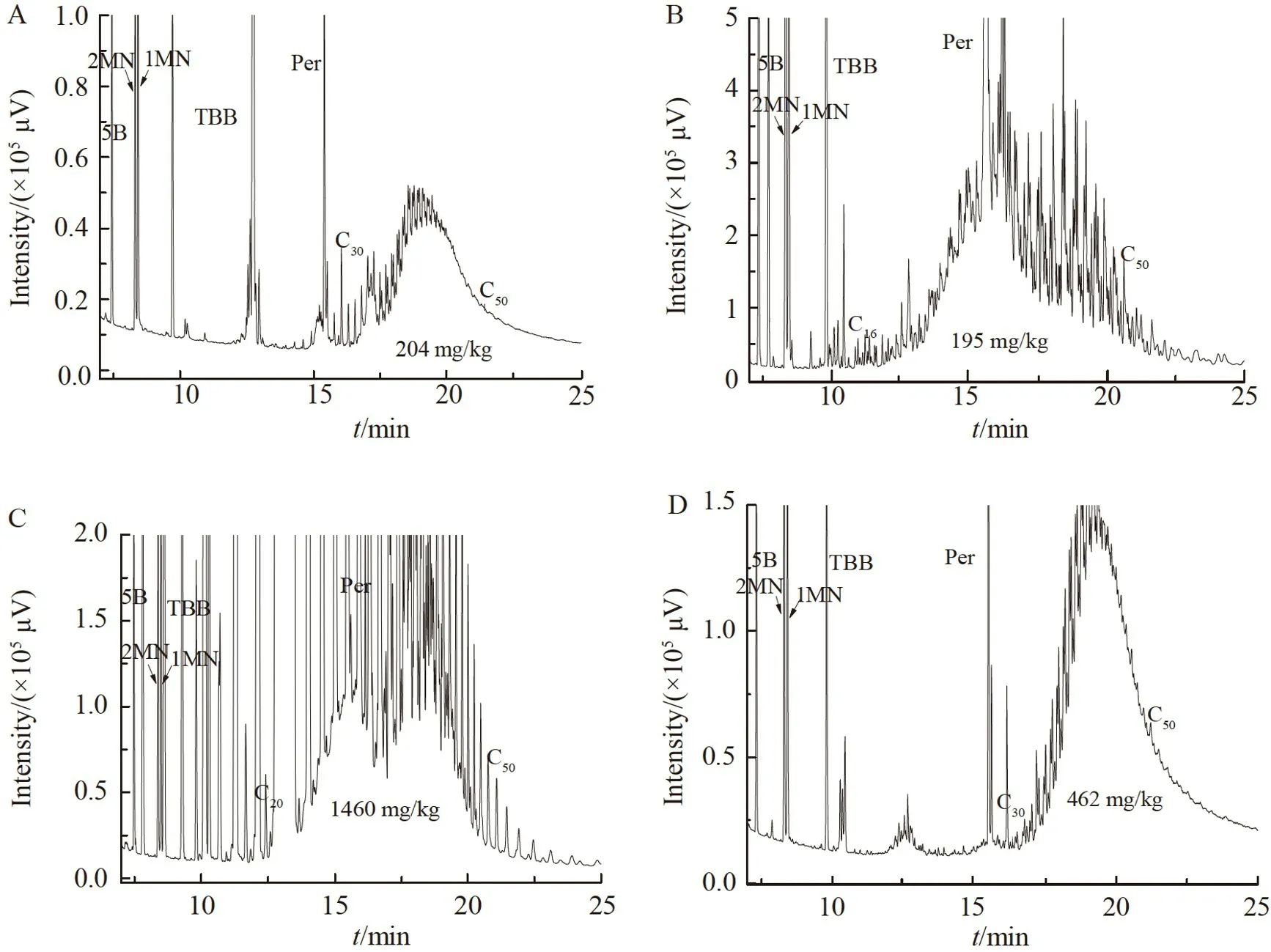
图2 4 个化妆品阳性样本的MOAH 谱图Fig.2 MOAH chromatograms of 4 positive cosmetic samples
3 结论
化妆品中微量MOAH 的分析难点在于与其共存的MOSH 干扰严重。本研究采用Prep-LC 成功实现了高达2 mg 的MOSH 与MOAH 的精准分离,结合LC-GC 联用技术建立了化妆品中MOAH 的高灵敏度检测方法。本方法的LOQ 低至10 mg/kg,可用于常规检测。采用本方法检测发现市售的部分口唇护理品中含有MOAH,其来源可能是精炼程度较低的润滑油或润滑脂原料。
支持信息
Supporting information
