基于跨视角相似度顺序保持的基因特征提取方法
2023-02-18苏树智张开宇王子莹张茂岩
苏树智 张开宇 王子莹 张茂岩
①(安徽理工大学计算机科学与工程学院 淮南 232001)
②(合肥综合性国家科学中心能源研究院(安徽省能源实验室) 合肥 230031)
1 引言
随着生物信息学的快速发展,人们对于癌症的研究已经发展到分子水平,脱氧核糖核酸(Deoxyribo-Nucleic Acid, DNA)微阵列技术[1]为人类在分子水平进行疾病诊断和治疗提供了全新手段,DNA微阵列技术可以大规模地快速检测基因表达情况获得基因表达数据,通过对基因表达数据进行分析可以了解细胞当前的生理状态区分癌变细胞与正常细胞,以便做出精准的诊断。基因表达数据样本个数通常为几十到几百,而每个样本的基因数量却成千上万,高维小样本作为基因表达数据的显著特性给大多数统计方法带来了挑战,对基因样本直接进行分类会存在维数灾难[2]问题,往往需要对基因表达数据进行维数约减,其目的是将原始数据投影到低维子空间以获得新的特征,该数据可以消除噪声和冗余信息利于后续处理。特征提取[3]作为最重要的维数约减方法之一,可以获得具有鉴别能力的特征,因此如何对关键有效基因进行特征提取成为基因分类研究的关键问题。
作为经典的单视角特征提取方法,主成分分析(Principal Components Analysis, PCA)[4]和线性判别分析(Linear Discriminant Analysis, LDA)[5]已经广泛应用于基因数据分析领域,Nakayama等人[6]使用基于高斯核的主成分分析方法用于基因表达数据聚类,讨论核参数的选择对于聚类性能的影响。Clayman等人[7]将PCA应用于研究DNA微阵列数据和临床变量之间的相互关系。Wang等人[8]提出了一种稀疏线性判别分析特征选择方法,与其他方法相比能够使用更少的特征或基因数量在降低错误分类率的情况下获得更好的结果。Lin等人[9]利用线性判别分析方法构造了阿尔茨海默病诊断框架,与之前研究中的方法相比,所提出的框架能够取得更好的分类性能。
随着信息的爆炸式增长,这种对于同一目标的一种表示的单视角学习方法已经不能满足研究者的需要,针对同一目标多种表示的多视角学习方法成为大势所趋,多视角学习既可以充分利用视角间的互补性,又能有效剔除视角间的冗余性,从而提取更具鉴别性的特征表示,在联合维数约减任务中,多视角数据可以发挥出比单视角数据更佳的识别性能。作为多视角学习的经典工具,典型相关分析(Canonical Correlation Analysis, CCA)[10]能够揭示两个不同视角之间的多元关系,CCA旨在找到一组基向量对,最大化从同一目标的两种不同视角获得的两个不同样本集之间的相关性,CCA已广泛应用于生物信息学领域,Lin等人[11]提出了组稀疏典型相关分析方法,引入组约束利用相关分析中的结构信息研究单核苷酸多态性与功能性磁共振成像测量的大脑活动之间的对应关系。Tenenhaus等人[12]提出了核广义典型相关分析方法,并提供了一个考虑块之间先验连接图的多块数据分析的通用框架对胶质瘤不同视角基因组数据进行分析。Wang等人[13]利用稀疏多元回归与稀疏典型相关分析之间的显式联系提出了基于特征向量的稀疏典型相关分析,研究甲状腺组织学图像和基因表达数据的相关性。
作为CCA的一种广义化扩展,多视角典型相关分析(Multi-view Canonical Correlation Analysis,MCCA)[14]能够对多个样本集之间的相关性进行表示,在不同研究中,多视角也通常被称为多模态或多重集等。MCCA中最佳线性变换可以通过求解广义特征值问题来获得,这对于高维数据来说计算量很大,样本的协方差矩阵也往往具有奇异性,这使得求解相关广义特征值问题具有挑战性。另外,MCCA只能以全局方式获得样本对之间的线性相关性,无法处理复杂的非线性情况。作为一种无监督的方法,MCCA没有利用监督信息,导致分类性能有限。
在过去的几十年里,MCCA为了解决这些限制已经扩展出了许多不同的新方法。当特征数量超过样本数量的情况时会导致协方差矩阵的奇异性,为了应对这一情况提出了正则化MCCA(Regularized Multi-view Canonical Correlation Analysis, RMCCA)[15]。利用正则化思想,通过图诱导嵌入多表示数据的几何结构信息构建了图正则化MCCA(Graph regularized Multiset Canonical Correlations, GrMCCs)[16],在人脸数据集上的应用表明GrMCCs方法能够获得较佳实验结果。核MCCA(Kernel Multi-view Canonical Correlation Analysis, KMCCA)[17]是MCCA的一种流行的非线性扩展,将原始非线性数据隐式映射到高维特征空间,使其具有线性可分性,从而在高维空间中执行线性典型相关分析方法。利用样本类别信息构建视角间的鉴别相关性,提出了鉴别型MCCA(Discriminative Multi-view Canonical Correlation Analysis,DMCCA)[18],使得子空间中具有紧密的类内分布以及类间离散分布,从而提高了低维特征的鉴别能力,在人类情感识别方面展现出良好性能。基于标签的MCCA(Labeled Multi-view Canonical Correlation Analysis, LMCCA)[19]充分利用训练样本的类内散布矩阵和多变量互相关矩阵来提取鉴别信息,建立了基于类内信息进行典型相关分析的统一框架,该方法在人脸识别和利用多重特征目标识别等应用验证了其有效性。
目前已有的各种多视角学习方法主要是通过不同的优化准则将多视角数据投影到子空间,从而保留原始数据的有效鉴别特征,但是在利用子空间进行学习时,往往忽略了投影前后样本之间的相似性[20],相似度顺序保持是一种重要的数据性质,它能够利用样本间的相似性来构建稳定的样本结构,因此本文提出一种相似度顺序保持特征提取方法,即相似度顺序保持跨视角相关分析(Similarity Order Preserving Across-view Correlation Analysis,SOPACA),SOPACA能够利用高维训练样本为每个视角学习到一组投影方向,通过将原始高维基因样本投影到相似保序子空间,从而获得更具鉴别力的相似性特征,通过构建鉴别敏感的视角内相似度顺序保持散布和约束鉴别敏感的视角间相似度相关,使得相似性特征在保持投影前后样本两两之间的相似性的同时具有类内聚集性与类间离散性,不仅保持了样本之间的结构关系而且充分利用样本监督信息。本文在肺癌及结直肠癌基因表达数据上进行针对性实验,实验结果表明本方法的优越性。
2 多视角典型相关分析

由于投影方向具有尺度不变性,MCCA方法可以表述为如式(2)所示优化问题

3 相似度顺序保持跨视角相关分析
3.1 构建鉴别敏感的视角内相似度顺序保持散布

利用相似度矩阵对式(3)进行化简,经过代数变换,式(3)能够推导为


3.2 构建鉴别敏感的视角间相似度相关

则式(1)中视角间相关性可以表述为



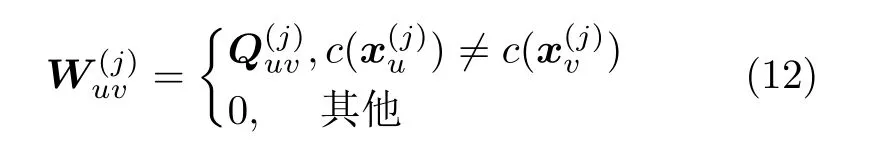
为了使得投影后相似保序子空间中不同类别的基因样本之间具有较大的类间距离,目标函数表示如式(13)所示


3.3 SOPACA的建模与求解
SOPACA方法期望学习的相关特征能够在保持不同视角间特征类内紧凑性和相似度顺序的同时具有较大的类间距离,这种期望可以表述为

通过最大化视角间相似度相关并且最小化视角内相似度顺序保持散布,将其与多视角典型相关分析的目标函数相融合,从而构建出相似度顺序保持跨视角相关分析方法,借助常用优化模型构造方法[21,22],SOPACA的优化问题可以描述为



4 实验结果与分析
为了验证SOPACA方法在癌症分类上的有效性,分别在肺癌和结直肠癌基因表达数据集上进行实验来评估SOPACA方法的识别性能。使用模态策略[24]获得基因表达数据的3种模态数据,具体而言,将基因表达数据看作时序信号,分别使用Coiflets,Daubechies和Symlets 3种小波变换提取其低频分量作为3种模态数据,由于基因表达数据具有高维与小样本之间的不平衡问题,使用PCA方法对模态数据维数统一约减至100维以保证实验的稳定性。实验中将SOPACA方法与LMCCA[19],GrMCCs[16], MCCA[14], LDA[5]方法进行对比分析,采用基于欧氏距离的最近邻分类器对基因样本进行分类得到最终识别结果。

算法1 SOPACA方法步骤
4.1 在肺癌基因表达数据集上的实验
肺癌基因表达数据集包含107个样本,每个样本均包含22283个探针测得的基因表达水平(下载地址为:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10072),其中癌症样本为58个,正常样本为49个。在实验中,随机抽取每类t(t=5,10, 15, 20, 25)个样本构建训练集,其余样本作为测试集,每个实验独立运行10次,最终得到如表1所示各算法在肺癌基因表达数据集上的平均识别率以及对应识别率的标准差。
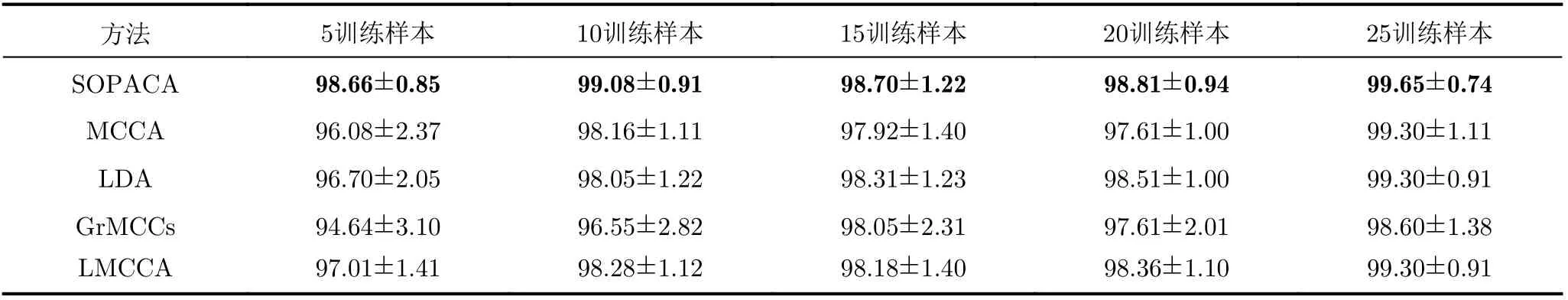
表1 在肺癌基因表达数据集上的识别率变化结果
MCCA只能全局地获取样本之间的线性相关性,因此在复杂的非线性情景下往往对数据拟合不足导致识别性能有限。LDA作为有监督的单视图学习方法,识别效果略优于MCCA方法。GrMCCs引入图结构考虑了多视角数据的几何结构,由于原始基因表达数据包含大量冗余信息及噪声,导致数据真实局部几何结构失真从而影响低维特征的鉴别力,这种失真关系使得GrMCCs的部分识别率低于MCCA。LMCCA充分利用样本类别信息获得了较高识别率,SOPACA方法保持了投影前后样本结构关系,充分利用类别信息获得类内紧凑性与类间分散性,随着样本数量增加SOPACA方法始终保持了最优识别率,在样本数量较小的情况下,SOPACA方法相较于其他算法显示出其优越性,对于具有高维小样本特点的基因表达数据尤为重要。标准差能够反映识别率的波动情况,标准差越大说明数据波动性越强,与其他方法相比,SOPACA方法拥有较小标准差表明识别率变化平缓,说明所提出的方法具有良好的鲁棒性。
4.2 在结直肠癌基因表达数据集上的实验
结直肠癌基因表达数据集包含34个样本,每个样本均包含54675个探针测得的基因表达水平(下载地址为:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32323),其中癌症样本和正常样本均为17个。在实验中随机抽取每类t(t=2, 3, 4, 5,6)个样本作为训练样本,其余样本用于测试,每个实验独立重复10次得到如表2所示各算法在结直肠癌数据集上的平均识别率以及对应识别率标准差。
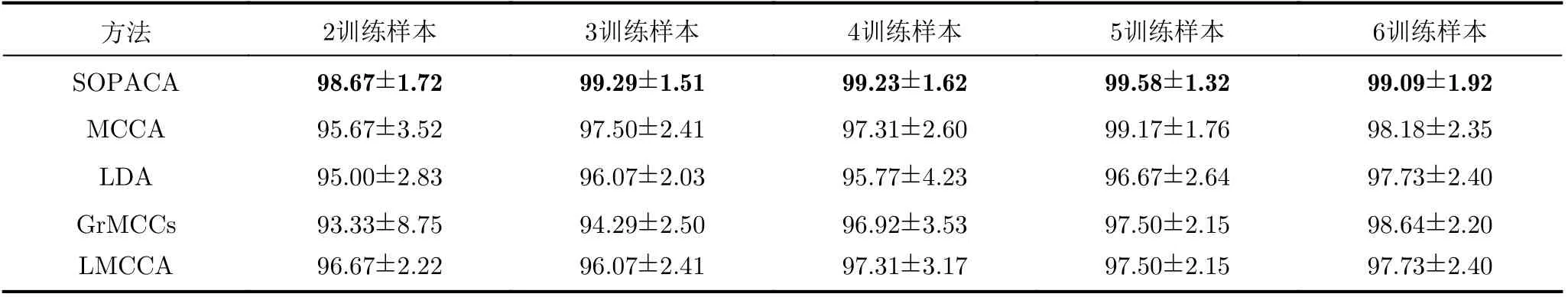
表2 在结直肠癌基因表达数据集上的平均识别率
MCCA方法只能处理简单的线性问题,无法提取更具鉴别力的特征。LDA表现出了较低识别率结果,反映了多视角学习方法对于特征抽取的优越性。与实验4.1中结果类似,利用图正则化技术的GrMCCs方法由于局部失真带来的影响导致平均识别率较低。DMCCA方法利用样本类别信息构建视角间的鉴别相关性,LMCCA充分利用训练样本的类内散布矩阵和多变量互相关矩阵来提取鉴别信息,二者都取得了较好的识别性能。SOPACA方法通过将样本投影到相似保序子空间能够获得更具鉴别力的特征,不仅保持了样本之间的结构关系而且充分利用样本类别信息,实验结果表明SOPACA方法的识别精度始终优于其他对比算法,当训练样本数量较少时更能体现出相似度顺序保持的优势。
5 结束语
传统基于子空间投影的多视角学习方法往往会忽略投影前后样本之间的相似性,进而影响多视角学习性能。本文提出了SOPACA方法,通过将基因表达数据投影到相似保序子空间,该子空间中的低维数据能够在保持投影前后样本相似度的情况下具有类内聚集性与类间离散性,从而有效增强低维数据的鉴别能力,在维持样本结构关系的同时充分利用了样本监督信息。在基因表达数据集上的实验表明,本文算法抽取的特征相较于其他特征提取算法更具鉴别性。
