肠道菌群与Toll样受体2在胰岛素抵抗中的作用①
2023-01-28刘新月郭豪常晓彤河北北方学院临床检验诊断学重点实验室张家口075000
刘新月 郭豪 常晓彤(河北北方学院临床检验诊断学重点实验室,张家口 075000)
肥胖和2型糖尿病(type 2 diabetes mellitus,T2DM)是目前全球普遍流行的代谢性疾病,其中,胰岛素抵抗是二者发生、发展的关键因素[1]。胰岛素抵抗即机体产生的正常剂量的胰岛素难以发挥作用,从而导致胰岛素维持血糖平衡的作用下降。低度炎症与胰岛素抵抗密切相关,而模式识别受体Toll样受体2(Toll-like receptor 2,TLR2)参与了炎症反应的发生与进展。肠道菌群受饮食等环境影响,其组成的改变可能是肥胖和T2DM中的炎症驱动者。本文将综述TLR2与肠道菌群对胰岛素抵抗的影响,并探讨其相关性。
1 TLR2与胰岛素抵抗
胰岛素是由胰岛β细胞分泌,维持葡萄糖稳态的一种重要激素。在肌肉组织中,当机体血糖升高时,胰岛素与其特定受体结合,胰岛素受体底物-1(insulin receptor substrate-1,IRS-1)发生酪氨酸磷酸化,继而结合并激活磷脂酰肌醇3-激酶(phosphati⁃dylinositol3 kinase,PI3K),促进磷脂酰肌醇4,5-二磷酸(phosphatidylinositol-4,5-bisphosphate,PIP2)转化为磷脂酰肌醇(3,4,5)-三磷酸(phosphatidyl inositol-3,4,5-triphosphate,PIP3),并招募3-磷脂酰肌醇依赖性激酶-1(phosphatidylinositol-dependent kinase-1,PDK-1),PI3K的活化与PDK-1可共同导致其下游靶点蛋白激酶B(protein kinase,Akt)的磷酸化,启动PI3K/Akt信号通路,发挥降低血糖作用[2]。此途径在肝脏组织中,胰岛素的降糖作用由IRS-2介导[1]。同时,胰岛素也是调节细胞生长、能量利用、线粒体功能、自噬、氧化应激、突触可塑性和认知功能的重要生长因子[3]。其促生长的作用由丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)介导。
Toll样受体(Toll-like receptors,TLRs)作为一种广泛表达的模式识别受体,能够识别并结合病原相关分子模式(pathogen-associated molecular patterns,PAMPs)和损伤相关分子模式(damage-associated molecular patterns,DAMPs),被激活后可诱导多种促炎因子和抗病毒因子表达,促进炎症应答[4]。TLRs为膜结合受体家族,存在于人类体内的有10种,即TLR1~TLR10,主要表达于树突状细胞、单核细胞和巨噬细胞等免疫细胞,也可表达于肠道等多种组织的上皮细胞和内皮细胞[5]。TLRs在代谢综合征的发病机制中发挥重要作用[6]。其中,TLR2被认为是胰岛素抵抗和代谢综合征的核心之一[6-7]。激活的TLR2活化下游信号通路的途径有两条:即髓系分化因子88(myeloid differentiation factor 88,MyD88)依赖途径和MyD88非依赖途径。MyD88依赖途径是除TLR3以外 所有TLRs信 号转 导的 共 用 途 径[8]。TLR2与相应配体结合后,通过MyD88依赖途径招募IL-1R相 关 激 酶-4(IL-1R-associated kinase-4,IRAK4)、IRAK1和IRAK2,后两者结合TNFR相关因子6(TNF receptor associated factor 6,TRAF6),TRAF6随后与转化生长因子β活化激酶-1(trans⁃forming growth factor-βactivated kinase 1,TAK1)结合,激活IKK、MAPKs,导致NF-κB信号通路的活化,并促进p38和C-Jun NH3末端激酶(c-Jun N-terminal kinase,JNK)磷酸化,释放促炎因子,进而直接导致IRS-1丝氨酸磷酸化,抑制下游胰岛素信号传导,启动下游炎症级联反应,诱发胰岛素抵抗(TLR2信号通路及其与胰岛素抵抗的关系如图1所示)[9]。另外,在一定条件下,TLR2还可协助TLR4与脂多糖(lipopolysaccharide,LPS)结合,引发一系列炎症反应,导致胰岛素抵抗发生。
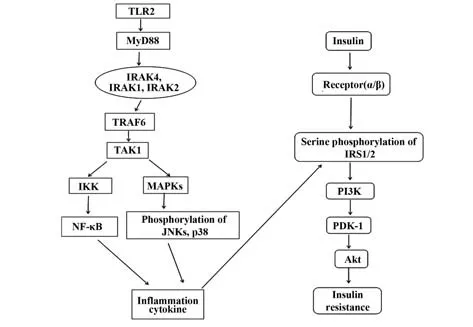
图1 TLR2信号通路与胰岛素抵抗的关系Fig.1 Relationship between TLR2 signaling pathway and insulin resistance
研究表明,T2DM小鼠骨骼肌细胞TLR2的表达明显高于正常小鼠[10];敲除TLR2基因可增加葡萄糖耐量和对胰岛素的敏感性,减轻胰岛素抵抗[11]。可见,TLR2信号通路活化导致机体处于低度炎症状态,进而影响正常胰岛素信号传导,可能是机体胰岛素抵抗的重要因素。
2 肠道菌群紊乱与胰岛素抵抗
健康成人体内的肠道菌群主要包括厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、梭杆菌门(Fusobacteria)、少数疣微菌门(Verrucomicrobia)、蓝藻菌门(Cyanobacteria)、螺旋体门(Spirochaeates)、VadinBE97菌门9个门和古生菌(Archaea)-史氏甲烷短杆菌,其中以厚壁菌门(G+)和拟杆菌门(G-)占比最大,约为90%。正常肠道菌群为维持人体健康所必需,但在某些异常或病理情况下,肠道菌群与人体这种共存关系遭到破坏,出现菌群失调,菌群失调会引起肠道通透性增加,促进炎症发生,进而导致胰岛素抵抗。既往研究表明,肥胖受试者的肠道菌群丰度降低,表现出更为明显的代谢紊乱和低度炎症[12-14]。肠道菌群的平衡可有效保护肠道,降低炎症与胰岛素抵抗发生的风险。肠道菌群失调导致胰岛素抵抗的发生目前有以下几个机制。
2.1 肠道菌群紊乱导致LPS升高促进胰岛素抵抗的发生LPS是革兰氏阴性菌表面细胞壁成分,是一种内毒素,可能与胰岛素抵抗的发生有关。有研究表明,以正常饮食喂养的小鼠持续皮下注射LPS 4周后,小鼠出现胰岛素抵抗[15]。肠道菌群中的拟杆菌能够改善肠道屏障功能,并降低血清中LPS浓度。当肠道菌群发生改变,如革兰阴性细菌拟杆菌裂解时,厚壁菌门与拟杆菌门比例升高,LPS被释放,出现血LPS明显升高,可引发强烈的免疫应答,产生炎症因子,以保护机体免受外来微生物感染。LPS作为TLR4的重要配体,可特异性激活TLR4。LPS与LPS结合蛋白(LPS-binding proteins,LBP)结合后,可进一步与CD14结合形成复合体,随后该复合体与TLR4/MD-2受体复合体结合,激活TLR4,继而激活MyD88依赖途径,启动NF-κB炎症信号通路,诱导产生炎症因子,抑制胰岛素信号通路,导致胰岛素抵抗的发生。
LPS除了可通过血液中单核巨噬细胞表面的TLR4激活全身炎症反应通路外,还可参与募集NOD样受体蛋白(NOD-like receptor proteins,NLRPs)、凋亡相关的斑点样蛋白(apoptosis associated specklike protein,ASC)和门冬氨酸特异性半胱氨酸蛋白酶-1(aspartic acid-specific cysteine protease-1,cas⁃pase-1)等炎症小体[16]。炎症小体是天然免疫系统的重要组成部分,在机体的免疫防御和疾病的发生中发挥重要作用。炎症小体的活化主要有两条途径:①通过与TLRs相互作用共同诱导NF-κB表达,进而促进促炎细胞因子IL-1β和IL-18的产生[17-18];②炎症小体(如NLRP3)能够对多种PAMPs和DAMPs发挥作用,识别多种细菌和组织损伤信号,激活并促进caspase-1依赖性炎症细胞因子IL-1β和IL-18产生[19]。炎症小体活化增加了炎症因子的数量,在一定程度上加速了胰岛素抵抗的发生。
2.2 高脂饮食诱发肠道菌群紊乱促进胰岛素抵抗的发生大量研究证实,高脂饮食(high fat diet,HFD)是导致胰岛素抵抗发生的重要因素之一,其中一个重要机制是导致系统LPS升高。研究表明,与低脂饮食喂养的小鼠相比,HFD喂养显著增加了LPS、IL-6、TNF-α和瘦素水平;同时,在门的水平上,HFD显著增加了变形菌门的相对丰度,降低了拟杆菌门的相对丰度,增加了硬壁菌和拟杆菌(F/B)比例,造成肠道菌群失调,从某种程度上导致了血清中LPS升高[20]。血清中LPS浓度过高容易导致机体发生代谢性内毒素血症,而肠道内的双歧杆菌可逆转代谢性内毒素血症,改善小鼠肠道完整性和相关代谢变化,长期高脂饮食则可能使双歧杆菌减少[15,20-21];同时,长期高脂饮食会导致肠道紧密连接相关蛋白claudin-1和闭合蛋白表达降低,破坏相关蛋白与肠道上皮细胞间构成的肠道屏障,肠黏膜的通透性增加,促进LPS渗出,为LPS释放入血提供条件;另外,LPS作为导致胰岛素抵抗的炎症因子,还可能通过浸润乳糜微粒或通过内皮细胞间连接的旁细胞通路运输进入血液循环,而高脂饮食可促进肠上皮细胞合成更多乳糜微粒,从而加速LPS吸收入血[22-23]。
高脂饮食导致胰岛素抵抗发生的另一个重要因子是瘦素抵抗。瘦素抵抗的原因可能有:瘦素中枢转运异常、内质网应激、受体信号传导发生障碍以及当血清瘦素水平增加时,机体可能出现高瘦素血症致其受体敏感性降低,引起瘦素抵抗。瘦素是一种由脂肪细胞分泌并释放入血的激素,其通过多种调节机制达到抑制食欲、控制脂肪生成和维持体重的作用。瘦素的作用机制主要有:①直接激活骨骼肌和肝脏组织中的5'-AMP-活化蛋白激酶(5'-AMP-activated protein kinase,AMPK)启动AMPK信号传导通路,促进脂肪酸氧化;②瘦素与其受体LRb结合为二聚体,通过激活JAK-STAT信号转导途径达到抑制食欲,减少能量摄取的作用;③通过直接作用于中枢神经系统发挥增加能量消耗、抑制脂肪生成、减轻体重的作用。高脂饮食致血清瘦素水平升高与肠道菌群失调有关。高脂饮食喂养的代谢综合征模型小鼠发生肠道菌群失调,出现厚壁菌门/拟杆菌门比例升高,同时加速脂肪细胞合成瘦素,瘦素信号传递减弱,小鼠出现肥胖以及瘦素抵抗[22,24]。一项针对无菌(germ-free,GF)小鼠的研究证实,肠道菌群缺失会导致瘦素表达和体质量增加,提示菌群紊乱会增加瘦素抵抗的风险[12]。
2.3 肠道菌群紊乱影响短链脂肪酸的代谢水平促进胰岛素抵抗的发生肠道是人体重要的消化器官,主要由小肠和大肠组成,由于小肠缺乏某些代谢酶导致人体内存在的许多碳水化合物和植物纤维不能被小肠消化,如多种膳食纤维、果胶、抗性淀粉以及不可消化的糖等,而大肠中的肠道菌群可对其进行发酵,产生发酵终产物短链脂肪酸(shortchain fatty acids,SCFAs)。肠道中较高浓度的SCFAs主要包括乙酸盐、丙酸盐和丁酸盐,其中乙酸盐占比最大。乙酸盐具有抗脂溶性作用,可减少脂质溢出到外周胰岛素敏感组织(如骨骼肌),这可能对改善胰岛素敏感性、维持葡萄糖稳态并降低下丘脑炎症具有重要作用[25]。丁酸盐能够刺激人肠上皮细胞TGF-β表达,进而激活抗炎调节性T细胞介导宿主免疫应答[26-27]。此外,SCFAs水平及丙酸盐和乙酸盐与胰岛素抵抗呈负相关[26,28]。SCFAs是肠道上皮细胞的主要能量来源,通过刺激肠道上皮细胞增殖及紧密连接蛋白的表达维持肠道屏障功能,有效阻止促炎因子LPS等进入血流,有助于减少全身炎症,逆转胰岛素抵抗。在T2DM中,患者出现SCFAs细菌数量下降,双歧杆菌数量下降,厚壁菌门/拟杆菌门升高,肠道通透性增加,血清LPS升高,机体出现内毒素血症、菌血症及低度慢性炎症等一系列症状[29-31]。对T2DM大鼠给予桑黄多糖提取物(phelli⁃nus linteus polysaccharide extract,PLPE)治 疗 后,SCFAs产生菌的丰度增加,使SCFAs水平提高,改善胰岛素抵抗[32];在饮食诱导肥胖小鼠中,补充SCFAs可改善胰岛素抵抗和肥胖[26,33]。因此,HFD时肠道菌群的紊乱影响了肠道内SCFAs等微生物代谢物的水平,继而影响糖脂代谢和能量代谢,促进脂肪堆积,刺激机体产生肥胖,诱发慢性炎症与胰岛素抵抗[32,34]。
3 TLR2、肠道菌群和胰岛素抵抗
慢性低度炎症与肥胖和T2DM患者糖代谢信号紊乱密切相关,代谢综合征患者外周循环系统中促炎细胞因子水平显著升高。前已述及,TLR2通过MyD88信号通路可诱导IL-1、IL-6和TNF-α等促炎细胞因子产生,参与胰岛素抵抗相关疾病,如肥胖和T2DM发展[35]。因此,TLR2表达和功能的改变及其细胞内信号通路的变化均可能影响胰岛素抵抗的发生和发展,而TLR2信号的活化依赖于TLR2和配体DAMPs或PAMPs的相互作用。
TLR2能识别内源性配体DAMPs,如透明质酸、β-抵抗素3、热休克蛋白、高迁移族蛋白B1等,其中有些是因为T2DM而被释放的[36];并且证实炎症是T2DM时胰岛B细胞损伤的主要原因[37]。因此TLR2和配体相互作用的炎症效果可能是T2DM进展的主要因素之一。有研究发现,T2DM患者TLR2基因表达上调,TLR2/MyD88/NF-κB信号通路的启动可加重机体炎症及胰岛素抵抗进程[38]。
肠上皮细胞是抵抗肠道微生物的第一道防线,拥有工作正常的免疫系统的完整肠壁是避免入侵所必须,而肠道微生物群与TLR在肠道的区域性表达有关。基于16srRNA基因的末端限制性片段长度多态性和克隆文库分析显示,与近端结肠和粪便相比,位于远端结肠的黏膜相关微生物群落的群落结构和多态性有显著差异,近端结肠与粪便聚类相似;对于SPF小鼠,结肠的近端与远端轴上TLR2和TLR4也有差异性表达,即TLR2在近端结肠中表达更高,向远端呈梯度下降,而TLR4在远端结肠中表达最高,向近端呈梯度下降;但在GF小鼠中未发现这种差异。因此,肠道微生物群对维持TLR的区域表达至关重要[39]。有研究表明,在正常情况下,肠道菌群能够通过TLR2、MyD88和PI3K途径激活肠道内产生IL-10的B细胞,这些B细胞能够减少结肠T细胞的活化,抑制攻击性免疫反应并维持黏膜内稳态,维持人体健康[40]。
肠道微生物群与宿主的相互作用是通过模式识别受体介导的,微生物群可直接与TLR相互作用并调节肠道免疫反应[41-42]。对小鼠使用广谱抗生素新霉素和杆菌肽可减少肠道中革兰氏阳性菌和革兰氏阴性菌,只留下少量未知菌,结果显示肠道菌群的减少可引起轻微炎症及回肠和结肠TLRs表达模式的显著变化,即显著增加TLR4表达,降低TLR2在回肠和结肠的表达,表明宿主-肠道微生物的相互作用发生改变[41]。
表达于天然免疫细胞和上皮细胞的TLR2能够检测PAMPs,肠道菌群是TLR2 PAMPs配体的重要来源之一。有研究表明,肠道菌群组成的变化增加了TLR配体输送到肝脏的数量,刺激肝细胞产生促炎细胞因子,故肠道菌群的组成变化是肝脏TLR信号激活的潜在触发因素[43]。TLR2可感知革兰氏阳性菌的细胞壁成分,如肽聚糖和脂磷壁酸;厚壁菌门是一种革兰氏阳性菌,也是肠道菌群的主要组成部分,在食用HFD的小鼠中,厚壁菌水平升高,表明TLR配体在肥胖小鼠的肠道菌群中极其丰富,TLR2表达也会相应增加[43]。
值得一提的是,本文重点阐述了肠道菌群及TLR2与T2DM的关系,T2DM表现为胰岛素相对不足,肥胖是导致T2DM的重要原因之一。糖尿病的另一个主要类型是1型糖尿病(T1DM),其是一种自身免疫性疾病,表现为胰岛素绝对不足,主要特征为T细胞介导的胰岛β细胞破坏,由遗传和环境因素共同引起[44]。肠道菌群紊乱是导致近年T1DM迅速攀升的重要因素之一,而环境因素对肠道菌群组成的影响远高于遗传因素[45]。肠道菌群紊乱时,肠道通透性增加,肠道毒素、食物抗原、感染因子可能从胃肠腔转移到肠道黏膜成分,最终转移到胰腺淋巴结,诱导胰岛β细胞凋亡,影响T1DM发展[46-47]。在T1DM动物模型中发现TLR2基因缺失对抵抗T1DM和胰岛炎有显著作用,TLR2通路的活化可加速T1DM进程[48]。
基于以上研究,在正常情况下,TLR2与肠道菌群相互作用共同维持宿主健康。但在菌群紊乱状态下,肠道菌群及其产生的代谢物活化TLR2,使TLR2表达发生变化,二者间的相互作用模式也发生改变,启动炎症应答,从而驱动肥胖和T2DM胰岛素抵抗的发生和发展;同时,肥胖和T2DM产生的DAMPs可进一步活化TLR2信号,加速疾病进程。
4 小结
炎症是糖代谢障碍的一个主要影响因素,而炎症应答的驱动者是紊乱的肠道菌群,其通过TLR2启动并促进炎症反应和胰岛素抵抗。然而,目前尚存在矛盾的结果,如GUADAGNINI等[49]研究发现,TLR2基因敲除小鼠厚壁菌门数量增加,蛋白细菌和拟杆菌数量降低,出现明显的胰岛素抵抗现象,矛盾结果出现的原因可能与动物饲养技术和环境不同有关,因为肠道菌群的组成可能主要受环境因素影响。在肠道菌群研究领域,利用基因敲除动物模型时,要特别注意研究技术和饲养环境。
肠道菌群紊乱活化TLR2信号与胰岛素抵抗密切相关,因此,靶向肠道菌群或炎症信号,通过平衡肠道菌群以及阻断TLR2信号通路的传导可能是治疗肥胖和T2DM胰岛素抵抗的有效途径。环境因素如饮食是改变肠道微生物组成的主要因素,膳食补充富含ω-3多不饱和脂肪酸的亚麻籽油可改善大鼠T2DM严重程度,其机制是通过调节肠道微生物群和抑制炎症,可见饮食干预肠道菌群有重要作用[50]。目前已经证实IL-1β参与T2DM胰岛β细胞损伤,TNF-α是外周胰岛素抵抗的关键介质,临床上IL-1β拮抗剂实验和抗TNF-α治疗可改善血糖水平、减轻T2DM及并发症症状,因此,靶向炎症的策略在T2DM患者的治疗中潜力很大[51]。肠道菌群紊乱状态下,可给予益生菌以平衡肠道菌群[31];通过粪便移植修饰现存的肠道菌群失调对改善个体应答和帮助调控炎症可能是一条新的有效路径[52]。
肠道菌群影响炎症反应和糖代谢的机制复杂,肠道菌群的研究是新兴的且具有挑战性的,但其研究潜力巨大。肠道菌群除了通过TLR2信号发挥作用外,可能与其他模式识别受体和炎症信号通路也存在交互作用。因此,尚需要进行进一步充分的基础与临床试验研究肠道菌群-TLR2及其他模式识别受体信号通路-胰岛素抵抗的相互关系与机制,以便更好地理解肠道菌群在炎症反应和胰岛素抵抗中的作用,从而为临床对T2DM等代谢性疾病提供准确的预防和诊疗靶点。
