玉米DUF1685基因家族的鉴定和进化分析
2023-01-16林正雨全津莹刘海岚
罗 璇,林正雨,陈 章,雷 波,李 洁,全津莹,刘海岚
玉米DUF1685基因家族的鉴定和进化分析
罗 璇1,林正雨1,陈 章1,雷 波1,李 洁1,全津莹1,刘海岚2*
1. 四川省农业科学院农业信息与农村经济研究所,四川成都 610066;2. 四川农业大学玉米研究所,四川成都 611130
本研究共鉴定出16个单、双子叶植物和卷柏的211个DUF1685基因家族成员。蛋白理化性质分析表明,211个基因的氨基酸序列长度为83~1071 aa,分子量为9.2~116.3 kDa,理论等电点为3.66~11.90。系统发育进化分析表明,DUF1685基因家族被划分了3个亚家族(Class I、Class II和Class III),Class I和Class II亚家族产生A1和A2、B1和B2的事件发生在单、双子叶植物分化后,同时A1和B2分支(成员均为双子叶植物基因)出现了基因扩张现象。保守基序和基因结构分析结果表明,同一亚家族成员之间保守基序相似,211个DUF1685基因家族成员中大部分含有1个内含子,少部分基因含有2个及以上内含子或者不含内含子。选择压分析表明,DUF1685基因家族在进化过程中相关位点受到正向选择,这可能是导致部分基因发生功能变化的原因。共线性分析表明,片段复制事件可能是玉米基因扩增和进化的潜在驱动力。基因启动子区域顺式元件分析结果表明,玉米基因启动子区域含有光响应元件、ABA和非生物胁迫相关的顺式作用元件,推测这些基因可能参与玉米对非生物胁迫的响应。转录组数据分析表明,玉米基因与特定组织(成熟花粉)的生长发育相关。本研究从多方面探究了玉米DUF1685基因家族的功能和进化情况,以期为今后深入研究玉米基因生物学功能和进化关系提供理论参考。
基因;基因鉴定;进化分析
未知功能结构域蛋白(domain of unknown function, DUF)是一群尚未被注释功能的蛋白[1]。已有研究发现,DUF基因家族成员广泛参与调控植物的生长发育,如DUF26亚家族的介导水稻根系发育并负调控根系卷曲[2],含有DUF647结构的与调控拟南芥早期幼苗形态和发育[3],DUF724基因家族成员通过RNA的转运参与植物细胞的极性生长[4],和编码的DUF642蛋白参与植物发育过程中果胶状态的精细调节[5]。同时,DUF基因家族成员也参与调控植物的花粉发育,如DUF784基因家族成员在介导胚囊发育的晚期或与花粉管的相互作用中起作用[6],含有DUF640结构域的基因控制水稻外稃和内稃发育[7]。此外,DUF基因家族成员也对各种非生物胁迫[8-10]和生物胁迫[11]积极响应。
玉米(ssp)是玉蜀黍属(L)一年生二倍体作物,起源于大约9000年前墨西哥野生大刍草[12]。玉米不仅是我国的关键农作物之一,也是人类生存的基本食物和动物饲料原料。面对全球气候的严峻变化,非生物胁迫对作物的危害也越来越严重。因此,可以充分利用玉米参考基因组测序信息[13],挖掘并研究与生长发育、胁迫应答等相关的未知基因,进而扩宽玉米基因资源。目前,随着各种植物基因组测序数据的公布,越来越多物种的DUF基因功能研究被报道与生长发育、胁迫响应有关,同时已有研究发现在甘蓝型油菜成熟花粉的转录组数据中含有DUF1685结构域的基因存在,但未对其基因功能进行深入探究[14],而DUF1685基因家族成员在其他植物中的功能研究还未见报道。因此,本研究基于已公布的16种单、双子叶植物和卷柏基因组数据,对DUF1685基因家族成员进行研究,包括成员鉴定、理化性质分析、系统发育树和选择压分析、蛋白保守基序和基因结构分析,全方位探究DUF1685基因家族的功能和进化情况,最终聚焦玉米DUF1685基因家族共线性分析和组织特异性表达,以期为今后深入研究玉米基因生物学功能和进化关系提供理论参考。
1 材料与方法
1.1 DUF1685基因家族鉴定和理化性质分析
从数据库Phytozome(http://www.Phytozome. net)下载甘蓝()、白菜()、菠萝()、拟南芥()、大豆()、柳枝稷()、棉花()、大麦()、苜蓿()、水稻()、高粱()和卷柏()基因组数据。从数据库Ensemble plant(http://plants.ensembl.org/index.html)下载甘蓝型油菜()和玉米()基因组数据。从数据库Brassica Database(http:// brassicadb.org/brad)下载芥菜型油菜()、黑芥()和萝卜()基因组数据。DUF1685(PF07939)的HMM模型从Pfam数据库(https://pfam.xfam.org/)获取。使用hmmsearch程序从下载的拟南芥蛋白序列中获取DUF1685保守的蛋白序列,然后以该序列为模板对下载的各物种的蛋白质序列进行blastp(E≤1e-9)搜索,所得候选序列使用Pfam(http:// pfam.xfam.org/search)、Smart(http://smart. embl-heidelberg.de/)和CDD(https://www.ncbi. nlm.nih.gov/cdd)软件去冗余。用在线分析软件ExPasy中的ProtPar am tool(http://web.expasy. org/protparam)分析基因蛋白序列的理化性质,包括分子量(molecular weight)和理论等电点(theoretical pI)。
1.2 DUF1685基因家族系统发育树和选择压分析
用Muscle 3.8.31软件对鉴定到的基因进行核苷酸多序列比对,然后使用MEGA v7[15]软件构建邻接发育树(NJ),重复抽样1000次。使用PAML[16](phylogenetic analysis by maximum likelihood)软件中的Site模型(M0、M1、M2、M3、M7、M8)计算dN/dS并确定正选择位点(经验贝叶斯法),并对M0/M3、M1/M2、M7/M8 3对似然比值进行似然比检验(likelihood ratio test, LRT)。
1.3 DUF1685基因家族蛋白保守基序和基因结构分析
使用MEME[17]软件分析所获候选DUF1685基因家族的蛋白保守基序,设置保守基序数目6个,保守氨基酸残基最小数目为10 aa,最大氨基酸数目为50 aa,其他参数默认。基因外显子和内含子结构使用Gene Structure Display Server 2.07[18]软件分析。
1.4 玉米DUF1685基因家族染色体定位和共线性分析
使用TBtools[19]软件可视化玉米基因的染色体定位。此外,使用带MCScanX[20]的TBtools软件分析玉米DUF1685基因家族的片段复制(fragment duplication)和串联重复(tandem duplication)事件。
1.5 玉米DUF1685基因顺式作用元件和组织表达特异性分析
使用Perl软件提取玉米基因CDS前2000 bp的上游序列,随后利用在线软件Plant CARE9(http://bioinformatics.psb. ugent.be/webtools/plantcare/html/)进行基因顺式作用元件预测,并使用在线网站DSGS(http://gsds.gao-lab.org/)进行可视化。在MaizeGDB数据库(https://maizegdb.org/)下载玉米‘B73’不同生长发育时期和部位的转录组数据,使用R[21]软件中的pheatmap包绘制聚类热图。
2 结果与分析
2.1 DUF1685基因家族鉴定
本研究在低等植物卷柏(),双子叶植物[拟南芥()、甘蓝型油菜()、甘蓝()、白菜()、萝卜()、芥菜型油菜()、黑芥()、苜蓿()、棉花()和大豆()]和单子叶植物[菠萝()、大麦()、水稻()、玉米()、柳枝稷()和高粱()]共17个物种中鉴定了211个基因(表1)。卷柏中有5个基因;10个双子叶植物中,基因成员数量由高到低排序为:甘蓝型油菜(34个)>芥菜型油菜(33个)>白菜=黑芥(16个)>萝卜=棉花(15个)>甘蓝(12个)>大豆(10个)>拟南芥(9个)>苜蓿(6个);单子叶植物中,DUF168基因家族成员数量由高到低排序为:柳枝稷(11个)>玉米(7个)>高粱=菠萝(6个)>水稻=大麦(5个)。蛋白理化性质分析表明,211个基因的氨基酸序列长度为83~1071 aa,分子量为9.2~116.3 kDa,理论等电点为3.66~11.90。

表1 DUF1685基因家族在植物中的分布
2.2 DUF1685基因家族系统发育进化分析
在构建的邻接树(NJ)中,DUF1685基因家族被划分了3个亚家族(图1):Class I、Class II和Class III,其中Class I包含2个分支A1和A2,Class II包含2个分支B1和B2。在Class I、Class II和Class III亚家族中分别有89、62和57个基因(图2A,图3A,图4A)。
在最大亚家族Class I中,分支A1的基因均是双子叶植物基因(67个,31.75%),分支A2中均是单子叶植物基因(22个,10.43%),表明在单、双子叶植物分化后产生了分支A1和A2,分支A1和A2分别含有67和22个基因,表明基因在分支A1中发生了基因扩张。在Class II亚家族中,分支B1的基因均是单子叶植物基因(11个,5.21%),分支B2中均是双子叶植物基因(51个,24.17%),表明也在单、双子叶植物分化后产生了分支B1和B2,分支B1和B2分别有11和51个基因,表明基因在分支B2中发生了基因扩张。

图1DUF1685基因家族系统进化树
2.3 DUF1685基因家族蛋白保守域和基因结构分析
使用MEME软件进行了蛋白保守基序分析,结构显示motif 1和motif 2为大部分基因所共有。除了这2个motif外,在Class I亚家族中(图2B),A1和A2分支主要包含motif 3。同时,A1分支大部分基因含有motif 5;而A2分支中单子叶植物除了菠萝含有motif 5,其余单子叶植物基因不含有motif 5,另外有小部分基因含有motif 6;在Class II亚家族中(图3B),主要包含motif 3,大部分基因含有motif 4和motif 6;在Class III亚家族中(图4B),所有的基因含有motif 4,部分含有motif 6。因此,结果表明Class II和Class III亚家族亲缘关系更近。
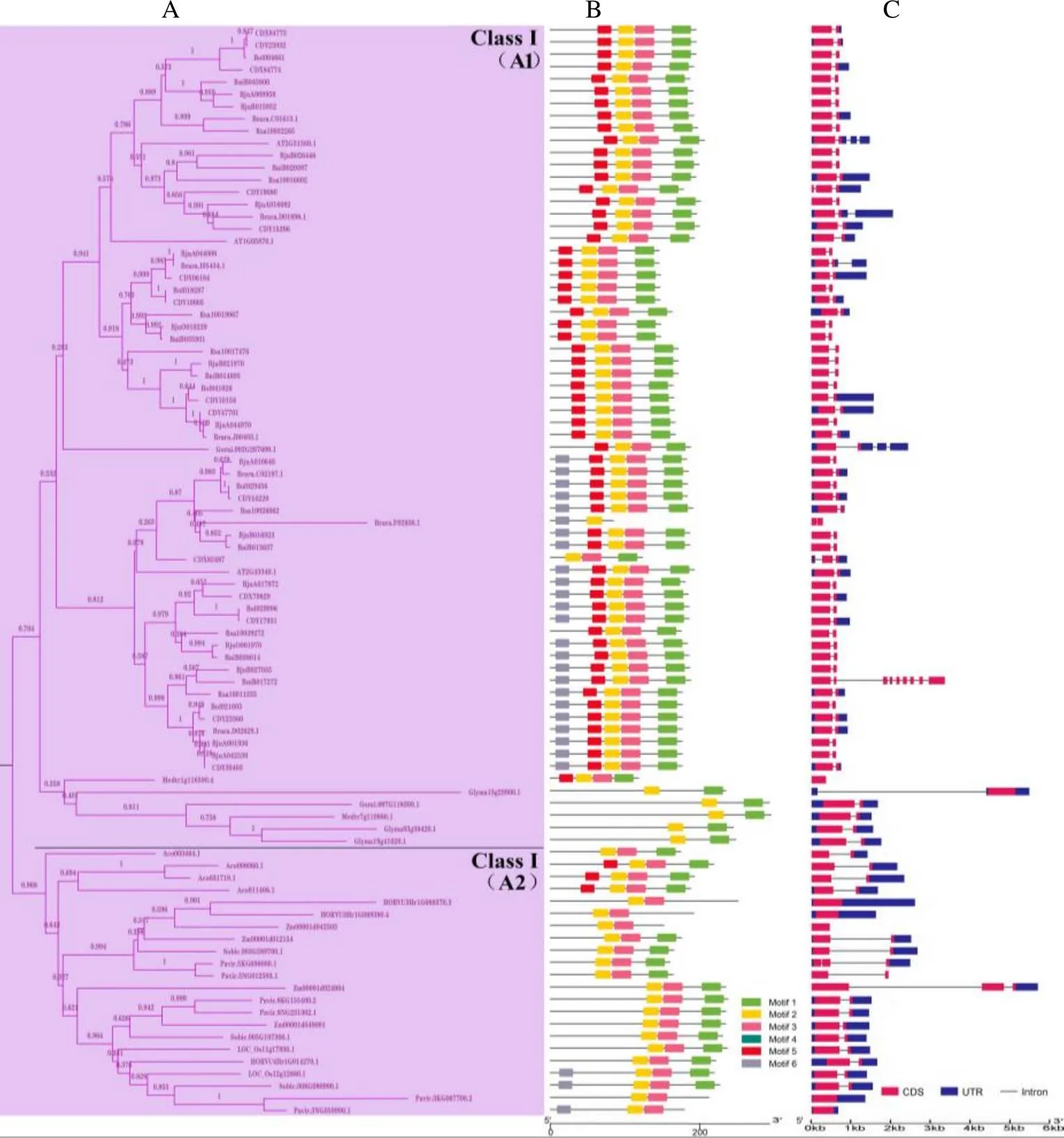
B:不同颜色代表不同保守基序;C:蓝色方块代表非编码区,红色方块代表CDS,灰色线条代表内含子。
B: Different colors represent different conserved motifs; C: The blue squares represent noncoding regions, the red squares represent CDS, and the gray lines represent introns.
图2DUF1685基因家族Class I亚家族系统进化树(A)、保守基序(B)和基因结构(C)分析
Fig. 2 Phylogenetic tree (A), conserved motif (B) and gene structure (C) analysis of DUF1685 gene Class I subfamily
基因结构分析结果显示,在Class I亚家族中(图2C),74个基因含有1个内含子,9个基因含有2个及以上内含子,6个基因不含内含子;在Class II亚家族中(图3C),54个基因含有1个内含子,6个基因含有2个内含子,2个基因不含内含子;在Class III亚家族中(图4C),42个基因含有1个内含子,11个基因含有2个及以上内含子,4个基因不含内含子。综上所述,在DUF1685基因家族中,大部分基因含有1个内含子,少部分基因含有2个及以上内含子或者不含内含子。
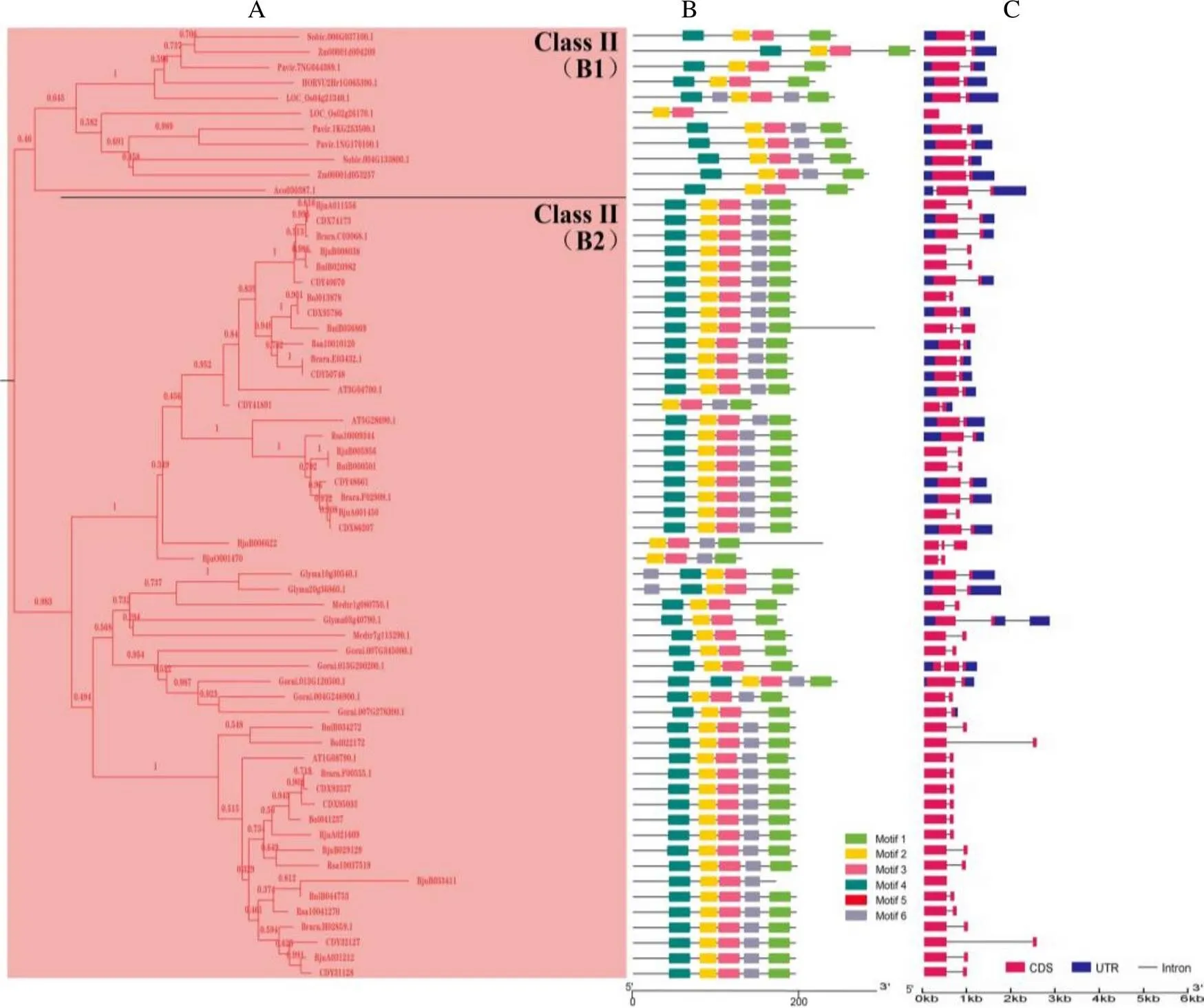
B:不同颜色代表不同保守基序;C:蓝色方块代表非编码区,红色方块代表CDS,灰色线条代表内含子。
B: Different colors represent different conserved motifs; C: The blue squares represent noncoding regions, the red squares represent CDS, and the gray lines represent introns.
图3DUF1685基因家族Class II亚家族系统进化树(A)、保守基序(B)和基因结构(C)分析
Fig. 3 Phylogenetic tree (A), conserved motif (B) and gene structure (C) analysis of DUF1685 gene Class II subfamily
2.4 DUF1685基因家族选择压分析
基于基因结构和保守基序分析,本研究发现不同亚家族不同基因之间存在差异,于是对各亚家族进行了选择压分析。运用CODEML软件中的Site模型计算了各亚家族的dN/dS值(表2)。A1分支中,似然比检验(LRT)显示Model M1和Model M2之间差异极显著(<0.001),因此Model M2分析证明,约有9.4%的位点受到正选择,ω2值为2.028 98;Model M7和Model M8之间差异极显著(<0.001),因此Model M8分析证明,约有23%的位点受到正选择,ω值为1.205 03。同时研究分析发现,A1分支在Model M2和Model M8分析得到的正选择位点均是129L。A2分支中,似然比检验显示Model M1和Model M2之间差异不显著(=1.000);Model M7和Model M8之间差异极显著(<0.001),因此Model M8分析证明,约有8.9%的位点受到正选择,ω值为7.676 29,分析得到的正选择位点是320和327。B1分支中,似然比检验显示Model M1和Model M2之间差异不显著(=1.000);Model M7和Model M8之间差异极显著(<0.001),因此Model M8分析证明,约有8.9%的位点受到正选择,ω值为113.620 63,分析得到的正选择位点是338P。Class III亚家族中,似然比检验显示Model M1和Model M2之间差异不显著(=1.000);Model M7和Model M8之间差异极显著(<0.001),因此Model M8分析证明,约有21.5%的位点受到正选择,ω值为42.241 90,受到正选择的位点有141个(表2)。综上所述,DUF1685基因家族的A1、A2、B1和Class III中基因部分位点遭受到了一定的正选择作用,推测DUF1685基因家族成员在进化过程中由于正选择作用,可能导致部分基因发生功能变化。
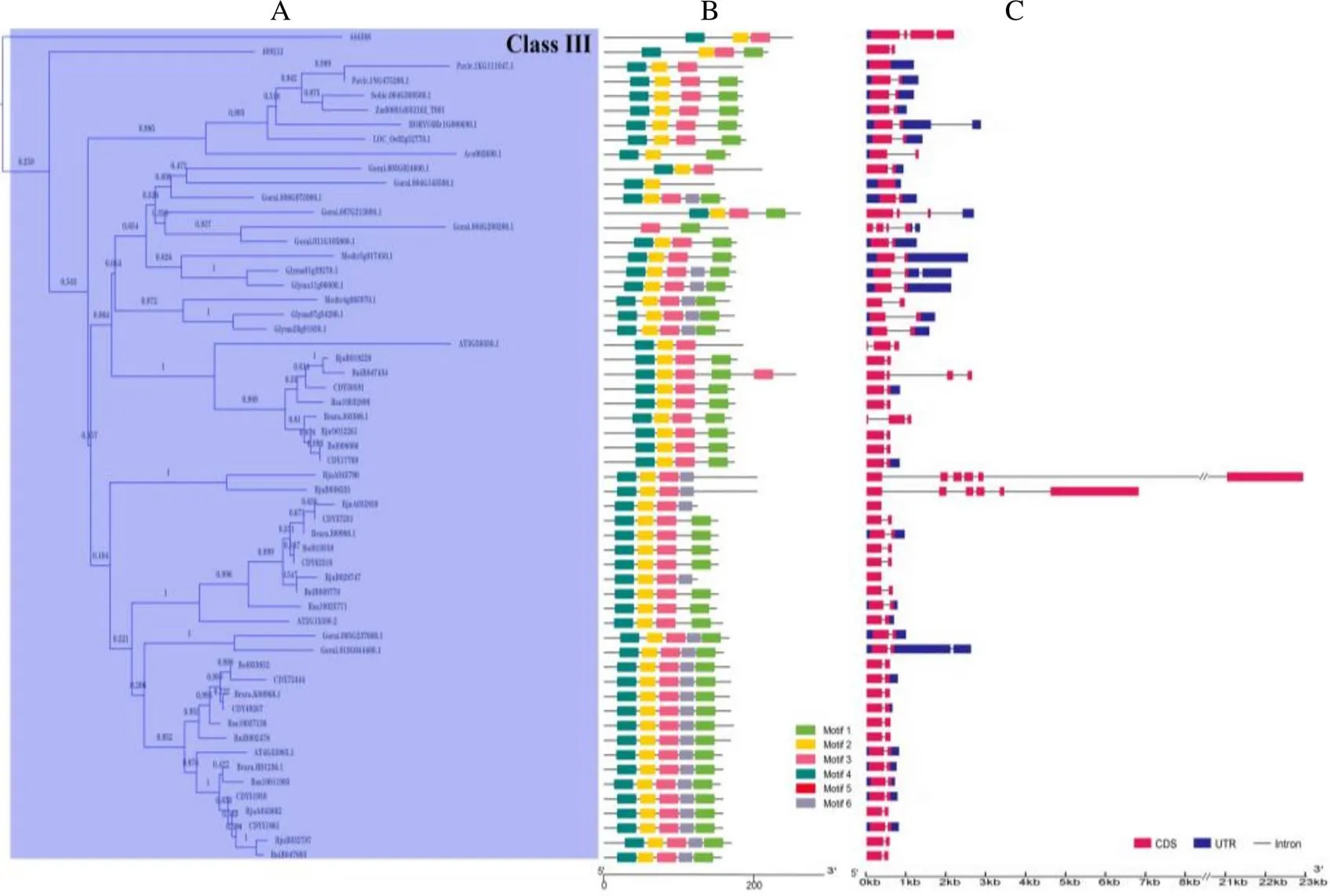
B:不同颜色代表不同保守基序;C:蓝色方块代表非编码区,红色方块代表CDS,灰色线条代表内含子。
B: Different colors represent different conserved motifs; C: The blue squares represent noncoding regions, the red squares represent CDS, and the gray lines represent introns.
图4DUF1685基因家族Class III亚家族系统进化树(A)、保守基序(B)和基因结构(C)分析
Fig. 4 Phylogenetic tree (A), conserved motif (B) and gene structure (C) analysis of DUF1685 gene Class III subfamily
2.5 玉米DUF1685基因家族染色体分布和共线性分析
利用玉米基因组注释信息和TBtools可视化了DUF1685基因家族的染色体分布。图5A结果表明7个在5条染色体上,每条染色体上的基因个数与染色体大小无关。最大的染色体(Chr01)不含基因,Chr02()、Chr03()、Chr08()和Chr 10()各含1个基因,Chr04含3个基因(、)。
全基因组复制分析对于物种的起源、进化和基因组扩展具有重要意义。因此,分析了玉米DUF1685基因家族复制事件,以了解基因复制事件的原因。结果显示(图5B),检测到1对基因有片段复制事件(segmental duplication events),Chr03的和Chr08的为片段重复基因对。此外,发现玉米基因不存在串联重复基因对事件。综上结果表明,基因的片段复制事件可能是玉米基因组中基因扩增和进化的潜在驱动力。
2.6 玉米DUF1685基因家族启动子序列分析
对启动子序列进行顺式作用元件预测(图6),发现7个玉米共有的顺式作用元件有7个。7个共有的顺式作用元件分别为ABRE、TATA-box、MYC、G-box、CAAT-box、MYB和STRE(表3)。7个顺式作用元件中,启动子常见顺式作用元件是转录起始–30核心启动子元件(TATA-box)和顺式作用元件(CAAT-box);与非生物胁迫相关的顺式作用元件包括脱落酸响应元件(ABRE)、干旱和ABA应答元件(MYB、MYC)、光响应元件(G-box)、热诱导元件(STRE)。由此可以推测,在玉米进化过程中,DUF1685基因家族成员在复制扩增时,依然有保守序列稳定选择,参与了玉米对非生物胁迫的响应。
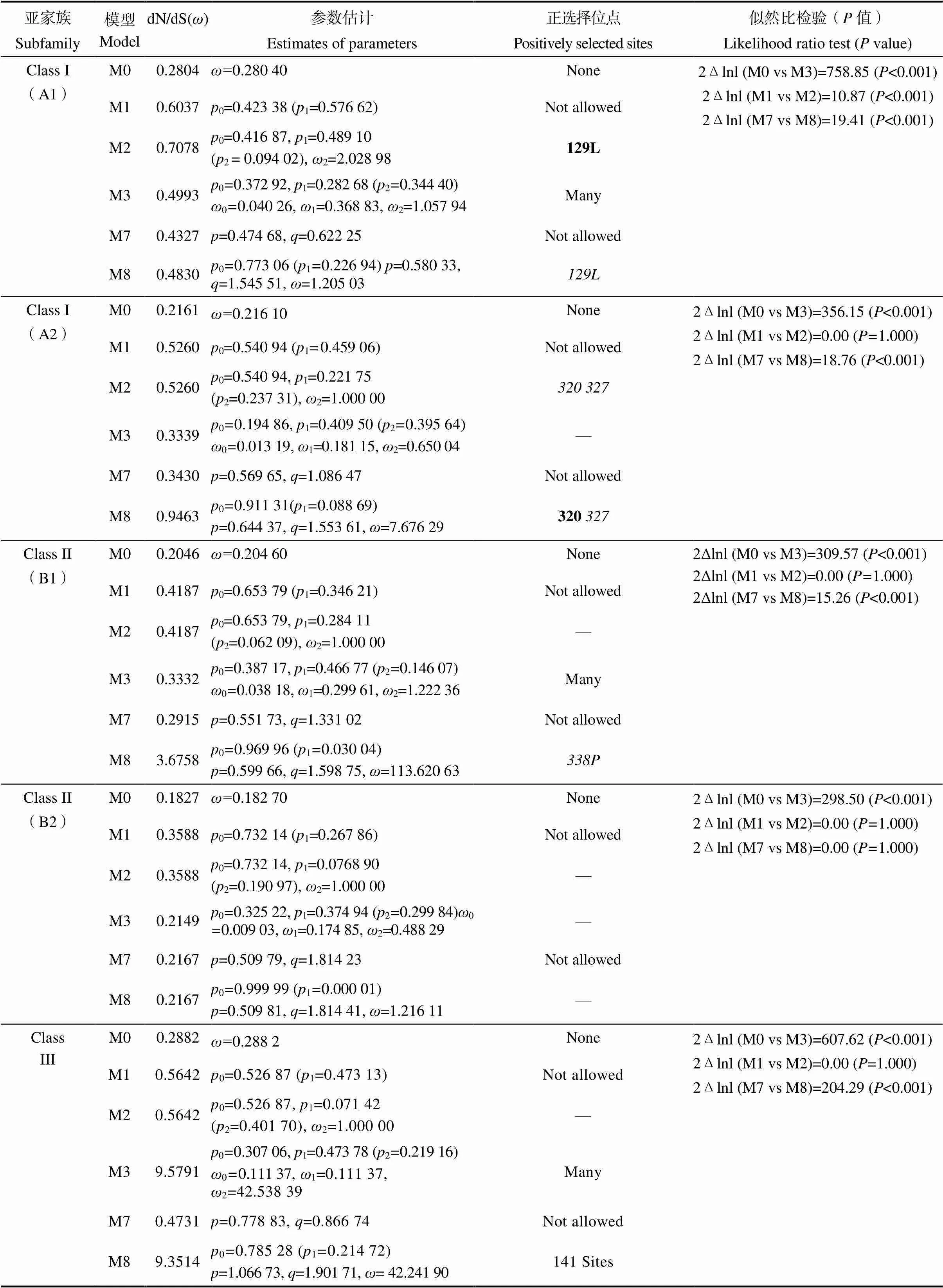
表2 Site模型下DUF1685基因家族dN/dS估计和似然比检验
注:在99%水平下推断为正选择的位点以粗体列出,95%水平的位点以斜体列出。
Note: Sites inferred to be under positive selection at the 99% level are listed in bold and those at the 95% level are in italic.
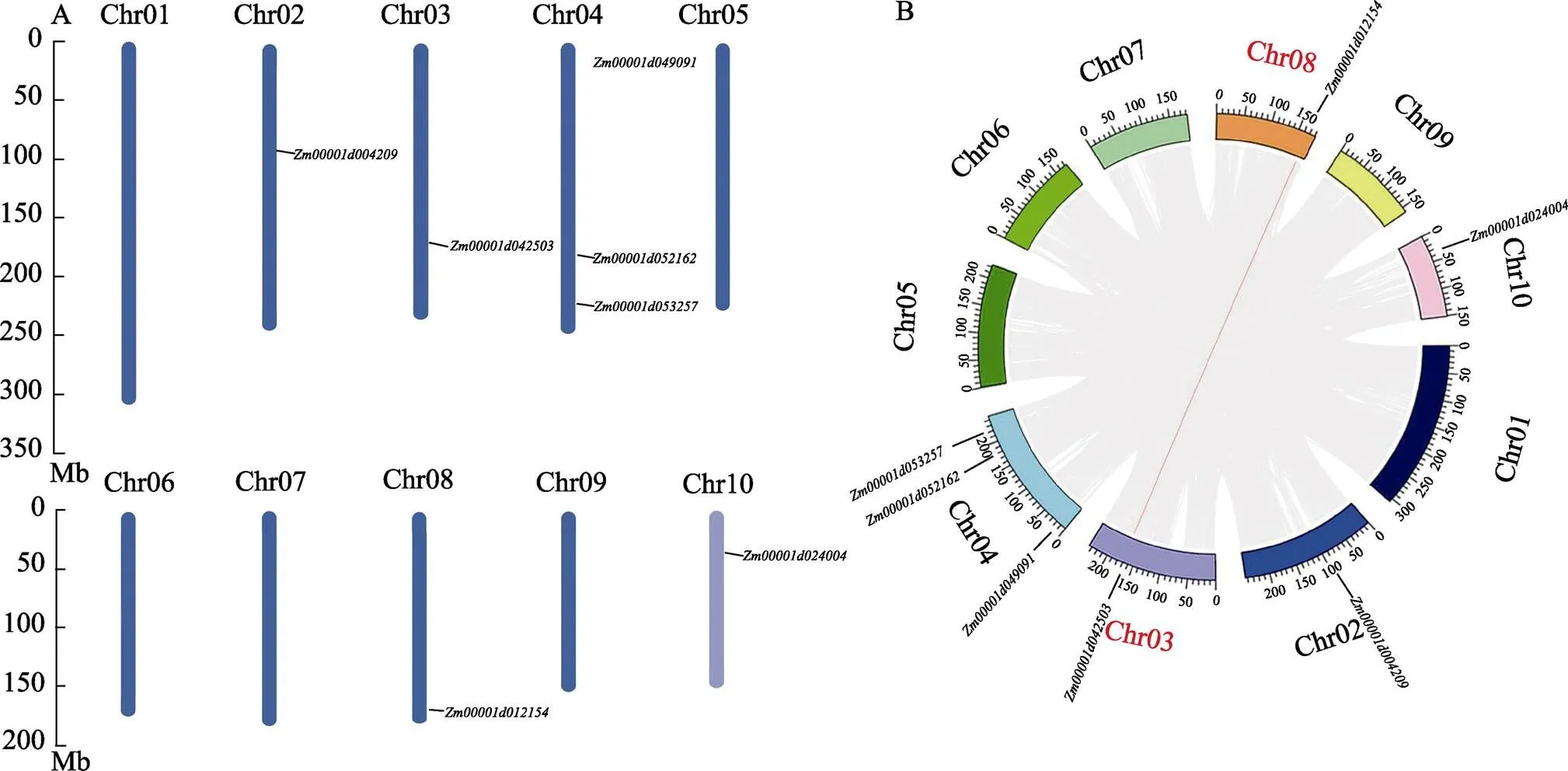
灰线代表玉米中所有基因的复制事件,红线代表DUF1685基因内的片段复制事件。
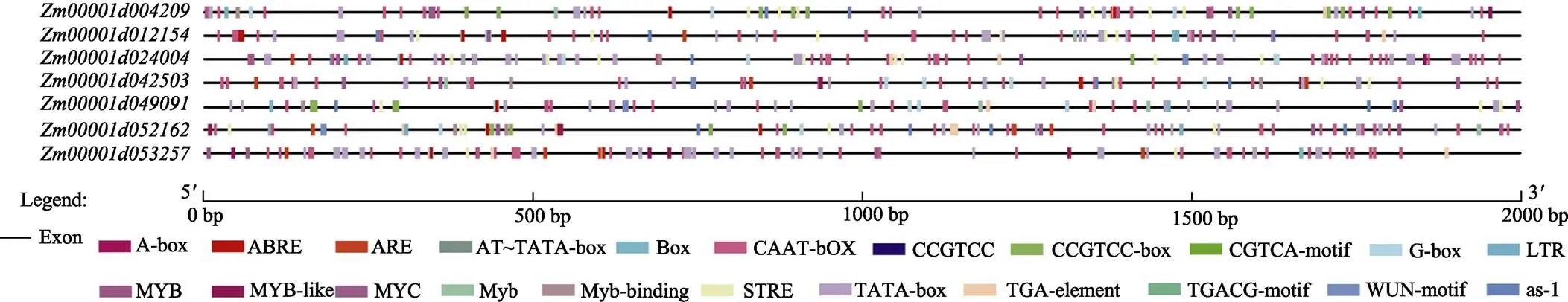
图6 DUF1685基因启动子顺式作用元件预测
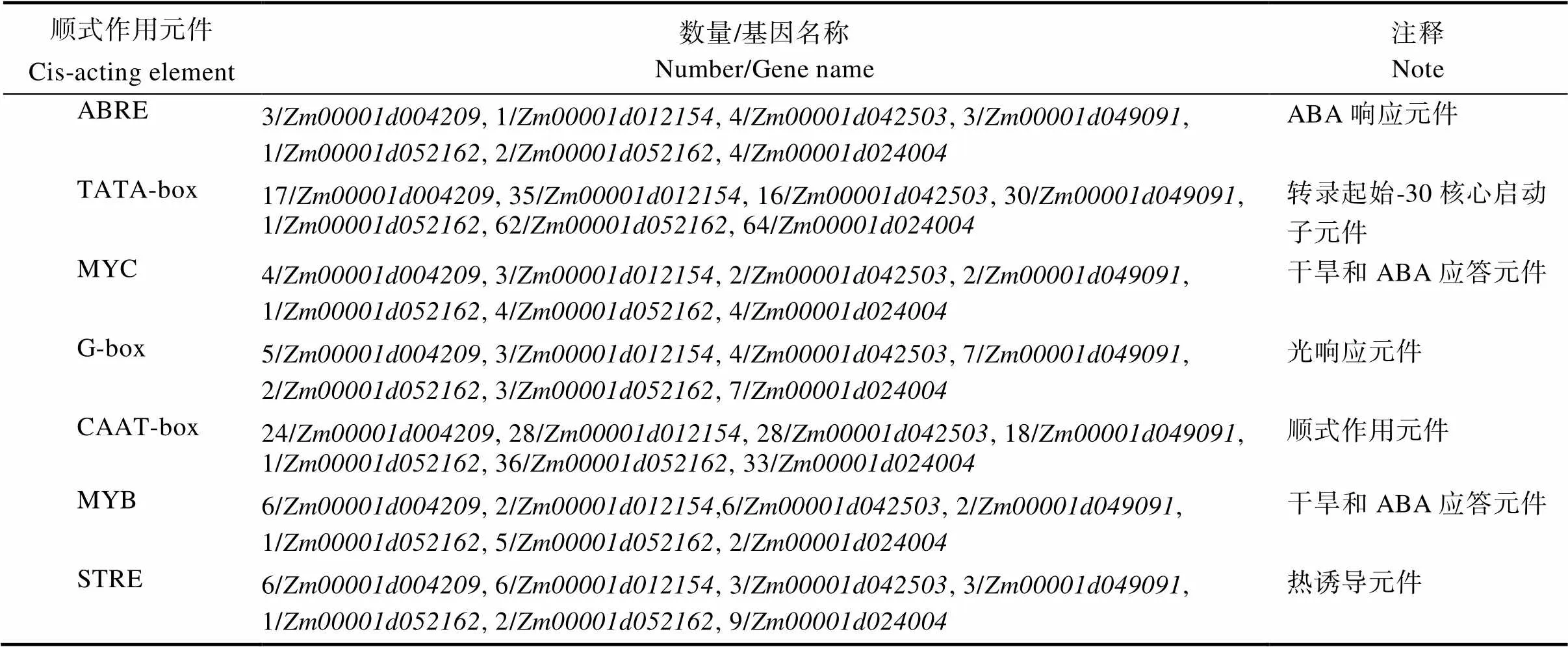
表3 玉米DUF1685基因启动子顺式作用元件分析
2.7 玉米DUF1685基因组织表达特异性分析
为探究玉米基因的时空表达特异性,对7个玉米基因()在玉米不同生长发育时期的转录组数据进行分析。基因表达聚类分析结果显示,玉米基因在不同组织中表达差异较大(图7)。在23个不同生长时期均未表达(未在表达聚类图7中显示),说明未参与玉米相关生命活动。和在玉米成熟花粉中表现出较高的表达水平;在玉米成熟花粉中表现出较低的表达水平。
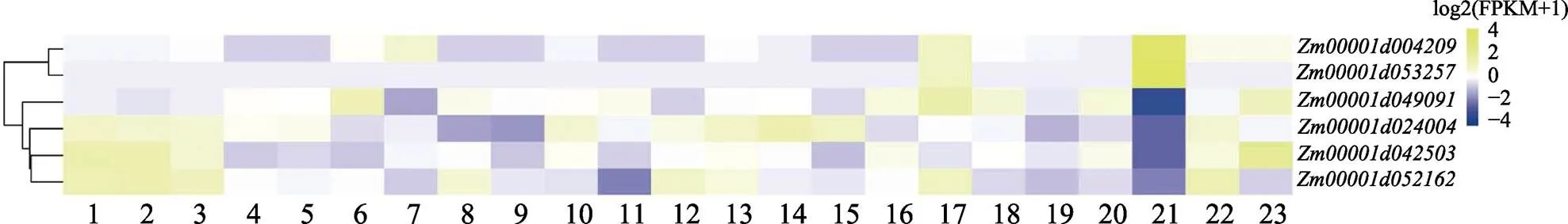
1:6~7节间;2:7~8节间;3:植物分生组织16~19 d;4:耳原基2~4 mm;5:耳原基6~8 mm;6:授粉后20 d胚胎;7:授粉后38 d胚胎;8:授粉后12 d胚乳;9:授粉后27 d胚乳冠;10:籽粒发芽后2 d;11:授粉后27 d果皮/糊粉层;12:叶区1(对称);13:叶区2(气孔);14:叶区3(生长);15:成熟的叶子;16:主根;17:根—皮质5 d;18:根—伸长区5 d;19:根—分生组织区5 d;20:次生根7~8 d;21:成熟花粉;22:雌性小穗当天收集为穗丝;23:穗丝。
3 讨论
3.1 通过理化性质分析DUF1685基因家族进化
目前,DUF1685基因家族的功能尚未被广泛研究。本研究利用生物学手段从16个单、双子叶植物和卷柏基因组中共鉴定到211个基因,各物种DUF1685基因家族成员数量不同以及各亚家族成员数量不同,一方面与各物种倍性不同有关,另一方面说明DUF1685基因家族存在一定的基因丢失或扩张现象。基因家族成员间的氨基酸数目、等电点、分子量区间范围较大,表明家族成员之间理化性质存在差异。同时,大多数基因家族成员具有相同的基因结构和保守基序,而各亚家族具有该亚家族特有的基因结构和保守基序,因此笔者认为DUF1685基因家族在进化过程中存在一定的保守性,同时在进化的过程中一些基因也发生了变化,以此来适应人工和自然选择。笔者认为通过分析基因理化性质,能在一定程度上揭示基因家族进化基本情况,为后续DUF1685基因家族进化深入分析奠定了基础。
3.2 顺式作用元件和基因表达对玉米DUF1685基因功能的影响
植物在生长发育过程中面临环境压力时,基因上游的顺式作用元件与特异性转录因子结合,特异性调节基因在植物中的表达,增加对环境的适应性,因此,启动子分析对研究基因功能起着至关重要的作用。MYB转录因子广泛分布于高等植物中,在植物抗逆反应中发挥着最重要的作用。已有研究表明,MYB转录因子通过与上游的顺式作用元件特异结合,参与植物次生代谢、激素和环境因子反应,在细胞分化、细胞周期和叶片形态发生中起重要调节作用[22-24]。拟南芥的脱水反应基因其启动子中一段67 bp序列对干旱和ABA应答,并且在这段序列上包括顺式作用元件MYC、MYB和GT-1[25]。G-box元件可能起到介导水稻叶片早期衰老诱导的表达,并且顺式元件的拷贝数显著影响表达水平[26]。本研究发现,7个玉米基因启动子均含有光响应元件(G-box),结合已有研究[26],说明玉米基因可能参与光合作用调控途径。在逆境胁迫方面,ABA响应(ABRE)、干旱响应(MYB和MYC)、热诱导调控(STRE)元件均存在于玉米基因中,说明基因在玉米应对这些逆境胁迫中均发挥着相应作用。基因在特定器官中高水平表达意味着其在相关生理过程中发挥重要作用。花粉发育过程中的基因,大致分为3类:第一类是组成型表达基因,第二类是花粉发育特异表达基因,第三类是介于前二者之间的在花粉中表达较高而在少数几种组织中也有少量表达的基因(玉米花粉发育相关基因的克隆及其原核表达)。学者通常认为只有在植物花粉发育过程中特异表达的基因,才能特异地在花粉发育中起作用[27]。ALLEN等[28]研究发现了1个结构保守,在花粉成熟晚期特异表达的基因。陈晓阳[29]研究发现,基因特异地在幼嫩的雄穗中表达,通过参与脂类物质的合成,从而调控花药角质层和花粉外壁的形成,进而影响花粉发育。在本研究中,玉米基因在成熟花粉中出现表达差异,暗示了其可能参与了花粉发育相关的生理活动过程,这为后续研究提供了玉米基因功能验证的初步方向。
3.3 选择压和片段复制与玉米DUF1685基因表达的关系
研究者通过检测基因组或基因中非同义与同义突变来研究自然选择对物种进化的影响。如果核苷酸序列的非同义替换率/同义替换率(dN/dS)的比小于1,则意味着纯净选择(purifying selection);如果比值等于1,则意味着中性选择(neutral selection);如果比值大于1,则意味着正向选择(positive selection)[30]。大部分茶叶基因[31]和向日葵HaWRKY基因家族在进化过程中都经历了强烈的纯化选择(dN/dS<1)[32]。在本研究中,7个玉米在进化过程中受到了正向选择,与茶叶基因和向日葵HaWRKY基因家族受到的选择压情况有所不同。同时,进化树结合基因表达情况分析发现,玉米成熟花粉中表现出高表达的2个基因(和)聚类在B1分支,低表达或零表达的4个基因(和)聚类在A2分支,说明序列相似度较高的基因可能其表达情况更相似。片段复制(segmentally duplicate)和串联重复(tandemly duplicate)是基因加倍的2种重要方式[33]。已有研究发现片段重复事件是杨树基因[34]和甘蓝型油菜基因[35]扩增和进化的力量。在本研究中,玉米DUF1685基因家族的1对片段复制基因(和)同属A2分支,而2个基因的表达情况存在一定差异。本研究仅下载了玉米‘B73’的转录组数据进行表达分析,后期有必要选择多个玉米品种和其他植物物种分析其表达情况。
[1] BATEMAN A, BIRNEY E, DURBIN R, EDDY S R, HOWE K L, SONNHAMMER E L. The pfam protein families database[J]. Nucleic Acids Research, 2000, 28(1): 263-266.
[2] JIANG J, LI J H, XU Y Y, YE H, BAI Y, ZHOU G X, LOU Y G, XU Z H, KANG C. RNAi knockdown ofroot meander curling gene led to altered root development and coiling which were mediated by jasmonic acid signalling in rice[J]. Plant, Cell & Environment, 2010, 30(6): 690-699.
[3] LEAURE C D, TONG H Y, YUEN G G, HOU X W, SEN X F, HE Z H. Root UV-B sensitive2 acts with root UV-B sensitive1 in a root ultraviolet B-sensing pathway[J]. Plant Physiology, 2009, 150(4): 1902-1915.
[4] CAO X, YANG K Z, XIA C, ZHANG X Q, YE C D. Characterization of DUF724 gene family in[J]. Plant Molecular Biology, 2010, 72(1-2): 61-73.
[5] ZúñIGA-SáNCHEZ E, SORIANO D, MARTíNEZ- BARA JAS E, OROZCO-SEGOVIA A, GAMBO-DEBUEN A., theDUF642 gene, is involved in pectin methyl esterase regulation duringseed germination and plant development[J]. BMC Plant Biology, 2014, 14: 338.
[6] JONES-RHOADES M W, BOREVITZ J O, PREUSS D. Genome-wide expression profiling of thefemale gametophyte identifies families of small, secreted proteins[J]. PLoS Genetics, 2007, 3(10): 1848-1861.
[7] LI X, SUN L J, TAN L B, LIU F X, ZHU Z F, FU Y C, SUN X Y, SUN X W, XIE D X, SUN C Q., a DUF640 domain-like gene controls lemma and palea development in rice[J]. Plant Molecular Biology, 2012, 78(4/5): 351-359.
[8] LUO C K, GUO C M, WANG W J, WANG L J, CHEN L. Overexpression of a new stress-repressive geneencoding a protein with a DUF966 domain increases salt and simulated drought stress sensitivities and reduces ABA sensitivity in rice[J]. Plant Cell Reports, 2014, 33(2): 323-336.
[9] HOU X, LIANG Y, HE X, SHEN Y. A novel ABA-responsivegene from wheat contributes to enhanced resistance to salt stress in[J]. Plant Molecular Biology Reporter, 2013, 31(4): 791-801.
[10] PALMEROS-SUáREZ P A, MASSANGE-SáNCHEZ J A, SáNCHEZ-SEGURA L,MARTíNEZ-GALLARDO N A, RANGEL E E, GóMEZ-LEYVAe J F, DéLANO-FRIER J P., an amaranth abiotic stress-induced DUF642 protein gene, modifies cell wall structure and composition and causes salt and ABA hyper-sensibility in transgenic[J]. Planta, 2017, 245(3): 623-640.
[11] XIE X Q, WANG Y J., a gene isolated from the Chinese grape, is involved in berry development and pathogen resistance[J]. Planta, 2016, 244(5): 1075-1094.
[12] MATSUOKA Y, VIGOUROUX Y, GOODMAN M M, SANCHEZ G J, BUCKLER E, DOEBLEY J. A single domestication for maize shown by multilocus microsatellite genotyping[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(9): 6080- 6084.
[13] SCHNABLE P S, WARE D, FULTON R S, STEIN J C, WEI F S, PASTERNAK S, LIANG C Z, WILSON R K. The B73 maize genome: complexity, diversity, and dynamics[J]. Science, 2009, 326(5956): 1112-1115.
[14] YIN L Q, ZHU Z D, HUANG L J, LUO X, LI Y, XIAO C W, YANG J, WANG J S, ZOU Q, TAO L R, KANG Z M, TANG R, WANG M L, FU S H. DNA repair- and nucleotide metabolism-related genes exhibit differential CHG methylation patterns in natural and synthetic polyploids (L.)[J]. Horticulture Research, 2021, 8(1): 142.
[15] SUDHIR K, GLEN S, TAMURA K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets[J]. Molecular Biology & Evolution, 2016, 33(7): 1870-1874.
[16] YANG Z H. PAML 4: phylogenetic analysis by maximum likelihood[J]. Molecular Biology and Evolution, 2007, 24(8): 1586-1591.
[17] BALEY T L, BODEN M, BUKE F A, FRITH M, GRANT C E, CLEMENTI L, REN J Y, LI W W, NOBLE W S. MEME Suite: tools for motif discovery and searching[J]. Nucleic Acids Research, 2009, 37(suppl_2): W202-W208.
[18] HU B, JIN J, GUO A Y, ZHANG H, LUO J, GAO G. GSDS 2.0: an upgraded gene feature visualization server[J]. Bioinformatics, 2015, 31(8): 1296-1297.
[19] CHEN C, CHEN H, ZHANG Y, THOMAS H R, XIA R. TBtools: an integrative toolkit developed for interactive analyses of big biological data[J]. Molecular Plant, 2020, 13(8): 1194-1202.
[20] WANG Y P, TANG H B, DEBARRY J D, TAN X, LI J P, WANG X Y, LEE T, JIN H Z, MARLER B, GUO H, KISSINGER J C, PATERSON A H. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity[J]. Nucleic Acids Research, 2012, 40(7): e49.
[21] TEAM D C R. R: A Language and environment for statistical computing[J]. R Foundation for Statistical Computing, 2011, 1: 12-21.
[22] CHEN S, PENG S, HUANG G, WU K, FU X, CHEN Z. Association of decreased expression of a Myb transcription factor with the TPD (tapping panel dryness) syndrome in[J]. Plant Molecular Biology, 2003, 51(1): 51-58.
[23] SUO J, LIANG X E, PU L, ZHANG Y S, XUE Y B. Identification ofencoding aMYB transcription factor that expressed specifically in fiber initials and elongating fibers of cotton (L.)[J]. Biochimica et Biophysica Acta (BBA)-Gene Structure and Expression, 2003, 1630(1): 25-34.
[24] YANG X Y, LI J G, PEI M, GU H, CHEN Z L, QU L J. Over-expression of a flower-specific transcription factor genecauses aberrant anther development[J]. Plant Cell Reports, 2007, 26(2): 219-228.
[25] IWASAKI T, YAMAGUCHI-SHINOZAKI K, SHINOZAKI K. Identification of a cis-regulatory region of a gene inwhose induction by dehydration is mediated by abscisic acid and requires protein synthesis[J]. Molecular & General Genetics, 1995, 247(4): 391-398.
[26] LIU L, XU W, HU X, LIU H J, LIN Y J.andelements play important roles in early senescence of rice flag leaf[J]. Science Report, 2016, 6: 20881.
[27] WILLING R P, MASCARENHAS J P. Analysis of the complexity and diversity of mRNAs from pollen and shoots of[J]. Plant Physiology, 1984, 75(3): 865-868.
[28] ALLEN R L, LONSDALE D M. Molecular characterization of one of the maize polygalacturonase gene family members which are expressed during late pollen development[J]. Plant Journal, 2010, 3(2): 261-71.
[29] 陈晓阳. 玉米雄性不育基因克隆与功能分析[D]. 北京: 中国农业大学, 2017.
CHEN X Y. Cloning and functional analysis of maize male sterility gene[D]. Beijing: China Agricultural University, 2017. (in Chinese)
[30] BOWERS J E, CHAPMAN B A, RONG J, PATERSON A H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events[J]. Nature, 2003, 422(6930): 433-438.
[31] PAUL A, CHATTERJEE A, SUBRAHMANYA S, SHEN G X, MISHRA N.gene family in:genome-wide identification, expression profiles, and regulatory network analysis[J]. Frontiers in Plant Science, 2021, 12: 777884.
[32] LI J, ISLAM F, HUANG Q, WANG J, ZHOU W J, XU L, YANG C. Genome-wide characterization ofgene family inL. and their expression profiles under biotic and abiotic stresses[J]. PLoS One, 2020, 15(12): e0241965.
[33] ZHU Y, WU N, SONG W L, YIN G J, QIN Y J, YAN Y M, HU Y K. Soybean () expansin gene superfamily origins: segmental and tandem duplication events followed by divergent selection among subfamilies[J]. BMC Plant Biology, 2014, 14: 93.
[34] ZHAO K, CHEN S, YAO W, CHENG Z H, ZHOU B R, JIANG T B. Genome-wide analysis and expression profile of thegene family in poplar[J]. BMC Plant Biology, 2021, 21(1): 122.
[35] CHEN Y, ZHOU R, HU Q, WEI W, LIU J. Conservation and divergence of the() genes related to flowering and circadian rhythm in[J]. Frontiers in Plant Science, 2021, 12: 760379.
Identification and Evolutionary Analysis of Maize DUF1685 Gene Family
LUO Xuan1, LIN Zhengyu1, CHEN Zhang1, LEI Bo1, LI Jie1, QUAN Jinying1, LIU Hailan2*
1. Agricultural Information and Rural Economy Research Institute, Sichuan Academy of Agricultural Sciences, Chengdu, Sichuan 610066, China; 2. Maize Research Institute, Sichuan Agricultural University, Chengdu, Sichuan 611130, China
A total of 211 DUF1685 gene family members in 16 monocotyledonous and dicotyledon andwere identified in this study. Analysis of protein physicochemical properties showed that the amino acid sequences of 211genes ranged in length from 83 to 1071 aa, molecular weights from 9.2 to 116.3 kDa, and theoretical isoelectric points from 3.66 to 11.90. Phylogenetic evolution analysis showed that the DUF1685 gene family were divided into three subfamilies (Class I, Class II and Class III). The events of A1 and A2, B1 and B2 in the Class I and Class II subfamilies occurred after the differentiation of monocotyledonous plants, and A1 and B2 branches (members were all dicotyledonous genes) had gene expansion phenomenon. The results of conserved motifs and gene structure analysis showed that the conserved motifs among members of the same subfamily were similar. Most of the 211 DUF1685 family genes contained one intron, and a few genes contained two or more introns or no introns. Selection pressure analysis showed that the related loci of DUF1685 family genes were positively selected during the evolution process, which may be the reason for the functional changes of some genes. Collinearity analysis indicated that fragment duplication events may be a potential driver ofgene amplification and evolution in the maize. The results of cis-element analysis in the gene promoter region showed that the promoter region of maizegene contained light-responsive elements, ABA and cis-acting elements related to abiotic stress. It was speculated that the genes may be involved in the response of maize to abiotic stress. Transcriptome data analysis revealed that the maizegene was associated with the growth and development of specific tissues (mature pollen). This study explored the function and evolution of the maize DUF1685 gene family in various aspects to provide a theoretical reference for the in-depth study of the biological function and evolutionary relationship of the maizegene in the future.
gene; gene identification; evolutionary analysis
S513
A
10.3969/j.issn.1000-2561.2022.12.005
2022-04-24;
2022-05-23
四川省重点研发项目(No. 2021YFYZ0028);四川省科技计划项目(No. 2020YJ0466)。
罗 璇(1995—),女,硕士,研究实习员,研究方向:作物遗传育种。*通信作者(Corresponding author):刘海岚(LIU Hailan),E-mail:lhlzju@hotmail.com。
