Metagenomic Analysis of Mangshan Pit Viper (Protobothrops mangshanensis) Gut Microbiota Reveals Differences among Wild and Captive Individuals Linked to Hibernating Behaviors
2023-01-05BingZHANGXiangyunDINGJianpingJIANGLinhaiLIandDaodeYANG
Bing ZHANG ,Xiangyun DING ,Jianping JIANG ,Linhai LI and Daode YANG*
1 Institute of Wildlife Conservation,Central South University of Forestry and Technology,Changsha 410004,Hunan,China
2 Qilu Normal University,Jinan 250200,Shandong,China
3 Chengdu Institute of Biology,Chinese Academy of Sciences,Chengdu 610041,Sichuan,China
4 Department for Wildlife and Forest Plants Protection,National Forestry and Grassland Administration,Beijing 100714,China
Abstract Gut microbiota play important roles in the immunity,digestion,and energy metabolism of their reptile hosts.Mangshan pit viper (Protobothrops mangshanensis) is a critically endangered snake species that is a Class I national protected species in China.Little is known regarding the relationship between P.mangshanensis and their gut microbial communities.In this study,the gut microbiota of wild P.mangshanensis individuals,artificially hibernating captive individuals,and non-hibernating captive individuals were compared across nine samples.Comparative shotgun metagenomic analysis was used to investigate the taxonomic composition,diversity,and function of P.mangshanensis gut microbial communities and assess whether their gut microbiomes were affected by their living environments and captivity conditions.The dominant phyla within P.mangshanensis gut microbial communities were Proteobacteria (65.55%),Bacteroidetes (15.97%),and Firmicutes (8.11%).Enriched functional pathways within the gut microbiota included metabolism (54.9%),environmental information processing (9.67%),and genetic information processing (9.37%).Wild snake gut communities exhibited higher microbial diversity than the other two groups.The gut microbiomes of wild and hibernating captive snakes may be more reflective of healthy intestinal homeostasis than that in nonhibernating snakes.Specifically,non-hibernating snakes exhibited increased levels of potentially pathogenic populations and functional specialization within gut microbial communities.Thus,different living environments and captivity methods may correspond to major shifts in microbiota composition,diversity,and function within P.mangshanensis.This study provides important insights to help guide the conservation of P.mangshanensis,while also carrying broad implications for our understanding of the effects of living environments and non-hibernating captivity conditions on the gut microbiota of snakes.
Keywords composition and diversity,gene functions and pathways,gut microbiota,metagenomics,Protobothrops mangshanensis
1.Introduction
Vertebrate guts are complex and dynamic systems that are inhabited by diverse and abundant microbial communities (Leyet al.,2008a;Leyet al.,2008b;Levinet al.,2021).Gut microbial communities and their hosts co-exist in mutually beneficial symbioses (Tonget al.,2020;Levinet al.,2021).Gut microbiomes can affect host physiology and behavior,while also regulating immune system activity and providing enzymes to enhance the host’s metabolic capacity (Shibataet al.,2017;Costeaet al.,2018).Indeed,a major function of the gut microbiota is the metabolic conversion of dietary and endogenous substrates that escape host digestion including carbohydrates,proteins,secondary plant metabolites,and xenobiotics (Costeaet al.,2018).
Animal gut microbial communities comprise two main structural components including: 1) core taxa that are putatively shaped by host genetics and have important physiological functions in hosts;2) the flexible pool that is acquired by contact with other individuals,through diets,or through coprophagy,or through interaction with other environments,which can promote the adaptation of hosts to new niches (Shapira,2016;Tonget al.,2020).Gut microbial communities are comprised mostly of bacteria,but also include archaeal,eukaryal,viral,and protozoal components (Kamadaet al.,2013).To date,most gut microbiota research of reptiles has focused on the bacterial components that are abundant and involved in the digestion and immunity of the host (Honget al.,2011;Keenanet al.,2013;Kohlet al.,2017;Zhanget al.,2019).However,little is known regarding other gut microbiota components in reptiles including eukaryal,archaeal,and viral community members.
Gut microbiota composition is affected by numerous factors,with the most important being the host’s evolutionary history and diet (Leyet al.,2008b;Youngblutet al.,2019).Gut microbiome studies have typically relied on captive animals that may have very different gut microbiome profiles relative to their wild counterparts (Kohlet al.,2014;Youngblutet al.,2019).Increasing studies have shown that the general impacts of captivity on animal gut bacterial communities can be assessed via changes in alpha and beta diversity analyses of gut bacterial communities (Kohlet al.,2014;McKenzieet al.,2017;Tanget al.,2020).For example,some studies have demonstrated declines in gut bacterial alpha diversity of captive individuals compared with wild counterparts (Kohlet al.,2014;Claytonet al.,2016).However,other studies suggest equivalent gut bacterial community alpha diversity among captive and wild individuals(Kohlet al.,2017;Eliadeset al.,2021),while a small number of studies also report increased alpha diversity in captive animal gut bacterial communities (Tanget al.,2020).In addition,many studies have documented significant differences in the beta diversity of gut microbial communities between captive and wild individuals (Kohlet al.,2017;Sandriet al.,2020;Eliadeset al.,2021),except for only a few comparisons (McKenzieet al.,2017).There are several mechanisms that may explain how captivity affects gut microbial communities including via influential factors like phylogenetic variability among hosts,the environmental sources of microorganisms,and host diets all representing important influential factors (Kohlet al.,2014;Trevellineet al.,2019a).
Hibernation is a typical example of extended fasting,and many animal taxa (including reptiles) undergo regular hibernation as part of their normal life histories (Dill-McFarlandet al.,2014;Sommeret al.,2016;Tanget al.,2019;Xiaoet al.,2019;Songet al.,2021a).Previous studies have shown that hibernation alters the composition and diversity of intestinal microbial communities.In addition,some hibernators exhibit restructured gut microbial communities during hibernation including the brown tree frog (Polypedates megacephalus),Chinese alligator (Alligator sinensis),ground squirrel (Ictidomys tridecemlineatus),and brown bear (Ursus arctos) (Dill-McFarlandet al.,2014;Sommeret al.,2016;Wenget al.,2016;Tanget al.,2019).During hibernation,animals exhibit slow metabolism,slow nutrient turnover,and greatly reduced body temperatures that may together lead to intestinal microbial communities being dominated by mucosal microbiota (Dill-McFarlandet al.,2014;Wenget al.,2016).Mucosal microbial communities that are closely associated with the mucus layer overlying the epithelium remain relatively stable over annual cycles and potentially serve as a pool for “seeding” microbial communities once exogenous substrates become available after hibernation(Zoetendalet al.,2002;Dill-McFarlandet al.,2014).In addition,mucosal microbial communities can exert greater regulatory effects on the expression of host immunity and epithelial functions relative to luminal microbiota (Schluter and Foster,2012;Dill-McFarlandet al.,2014).Consequently,hibernators are often accompanied by remodeled intestinal immune systems that are also aspects of host biology that are both influenced by,and can itself influence,microbial communities (Careyet al.,2013;Tanget al.,2019).
Gut microbial community studies have primarily focused on birds and mammals,with relatively few studies of gut microbial communities in reptiles (Tanget al.,2019;Tanget al.,2020).Nevertheless,limited studies have evaluated the effects of diet (Kohlet al.,2016;Jianget al.,2017;Montoya-Ciriacoet al.,2020),host genetics (Yuanet al.,2015;Renet al.,2016;Zhanget al.,2019),captivity (Sandriet al.,2020;Tanget al.,2020),gut region(Colstonet al.,2015;Kohlet al.,2017),disease (Ahasanet al.,2018;Linet al.,2019),gestation (Trevellineet al.,2019b),hibernation(Tanget al.,2019),geographic location (Scheelingset al.,2020),and altitudinal habitat gradients (Zhanget al.,2018) on the gut microbial communities of reptiles.Most of these studies have evaluated gut microbial community taxonomic profiles by sequencing bacterial 16S rRNA gene amplicons from the gut contents or fecal samples from reptiles (Colstonet al.,2015;Trevellineet al.,2019b;Zhanget al.,2019).In addition,a few studies have investigated the functional profiles of reptile gut microbiomes using shotgun metagenomic sequencing (Tanget al.,2019).
The Mangshan pit viper (Protobothrops mangshanensis) is a member of the reptile group and is the largest species of Viperidae in China (ranging up to 2 m long and 2–4 kg in weight),but has a very limited distribution,only occupying~10,500 ha (Gonget al.,2013;Yanget al.,2013).Mangshan pit vipers comprise a population estimated to be less than 500 individuals (Gonget al.,2013).They were consequently classified as an endangered species on the International Union for Conservation of Nature (IUCN) Red List of Threatened Species and as a critically endangered species on the Red List of China’s Vertebrates (Jianget al.,2016;Wang,2021),in addition to being listed in Appendix II of the Convention on International Trade in Endangered Species (CITES) of Wild Fauna and Flora in 2019.Further,P.mangshanensiswere listed as a Class I national protected species in China in 2021.The Hunan Conservation Center for Mangshan pit viper (HCCMP) was established to increase captive Mangshan pit viper populations,breed newborn individuals,and reintroduce Mangshan pit vipers into the field.At the HCCMP,P.mangshanensisare raised either with hibernation or without hibernation.Artificial hibernation refers to Mangshan pit vipers hibernating in artificial environments during the winter,while non-hibernating Mangshan pit vipers are active year-round without hibernation.Non-hibernating rearing requires raising the temperature of breeding houses in autumn and winter so that captive snakes move and eat normally.Non-hibernating rearing can lead to overwintering survival rates of youngP.mangshanensisthat exceed 80%,with the average weight gain of youngP.mangshanensisalso exceeding 50% (Chenet al.,2013).Consequently,this breeding method has been widely adopted for rearing snakes (Huet al.,2013;Liet al.,2013).However,non-hibernating Mangshan pit viper populations exhibit mysterious deaths during breeding,leading to decreased population numbers (Chenet al.,2013).In contrast,hibernating Mangshan pit vipers remain in good health and have stable populations.Although gut microbiota dynamics are clearly related to host gut health (Cerf-Bensussan and Gaboriau-Routhiau,2010;Sharpton,2018),the effects of artificial hibernation and non-hibernation captivity conditions on Mangshan pit viper gut microbial communities remain largely unknown.
Most gut microbiome studies have focused on the microbiota of birds and mammals,whereas little is known about the gut microbiota of reptiles,especially snakes (Tanget al.,2019;Smithet al.,2021).Here,metagenomic analysis was used to assess the composition,diversity,and function ofP.mangshanensisgut microbiota from wild,artificially hibernating,and nonhibernating individuals to comprehensively analyze the gut microbial communities ofP.mangshanensisheld in various states of captivity.We hypothesized that: 1) the composition,diversity,and functions of gut microbiota in wild snakes would differ from those of captive snakes;2)P.mangshanensisthat undergo artificial hibernation in captivity may be able to better regulate their gut microbiomes and maintain gut homeostasis;3) non-hibernating captive individuals will be more enriched in pathogenic microorganisms and microbiota with energy metabolism pathways.
2.Materials and Methods
2.1.Sample collectionP.mangshanensisare an extremely endangered species and their defecation behaviors are not well known (Jianget al.,2016;Wang,2021).In addition,the numbers of Mangshan pit vipers in captivity reared under hibernating and non-hibernating conditions are limited.To ensure consistency of sampling times and allow statistical inference among groups,only three samples from three different individuals were collected for each group.Specifically,three fecal samples were taken from wildP.mangshanensisand were collected from the Hunan Mangshan National Nature Reserve in June 2018.In addition,three fecal samples were collected from non-hibernating Mangshan pit vipers,and three were taken from hibernating Mangshan pit vipers at the HCCMP in July 2018.Feces were immediately sterilely sampled after defecation from wild Mangshan pit vipers (i.e,the WL group),non-hibernating Mangshan pit vipers (NHB group),and hibernating Mangshan pit vipers (HB group).The feces were frozen in liquid nitrogen and transported to the laboratory on dry ice,followed by maintenance at −80°C.After all sample collection,they were transported to Personal Biotechnology Co.,Ltd.(Shanghai,China) on dry ice for subsequent analyses.
WildP.mangshanensiswere born and entirely live in the wild,while the captive individuals were housed at the HCCMP.The large size of Mangshan pit vipers,slow movement speed,arboreal nature,and their defecation on the ground allow for easy observance at a distance and efficient sampling after defecation.Both hibernating and non-hibernating Mangshan pit vipers were born at the HCCMP in 2014 and were all raised in animal houses.The animal house for hibernating Mangshan pit vipers was maintained separately from non-hibernatingP.mangshanensis.Further,each snake was maintained separately in different feeding boxes.To ensure cleanliness of feeding box environments,they were cleaned every day,and the feeding box was replaced and disinfected every other month.To reduce environmental contamination of samples,the date of snake defecation (generally the third day after eating) was calculated and observations were regularly conducted on that day.In addition,to ensure the consistency of sampling times,feces were collected from both hibernating and non-hibernating snakes after the same feeding event.The temperature and humidity of the hibernating Mangshan pit viper enclosures varied with ambient conditions.Mangshan pit vipers hibernate when temperatures fall below 12°C.Consequently,the enclosures were maintained between 5–12°C during hibernation,which extended from November to April of the next year.Nonhibernating Mangshan pit vipers were kept at relatively stable temperatures and humidity levels year-round (27–30°C and 85–95%,respectively).Both hibernating and non-hibernatingP.mangshanensiswere fed on artificially raised small white mice (Mus musculus) every 15 days,whereas hibernatingP.mangshanensisdid not eat during hibernation.
This study was performed in accordance with the recommendations of the Institution of Animal Care and the Ethics Committee of the Central South University of Forestry and Technology (permit number: CSUFT#797217).Permission for fieldwork was obtained from the Forestry Department of Hunan Province (permit number: HNLY-5319171).
2.2.DNA extraction,library preparation,and sequencingTotal microbial genomic DNA was extracted from fecal samples using the DNeasy PowerSoil Kit (QIAGEN,Inc.,Netherlands) according to the manufacturer’s instructions,followed by storage at −20°C prior to further use.The concentration and purity of extracted DNAs were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific,Waltham,MA,USA) and agarose gel electrophoresis,respectively.Extracted DNA was used to construct metagenome shotgun sequencing libraries with insert sizes of 400 bp using the Illumina TruSeq Nano DNA LT Library Preparation Kit.Sequencing was performed on the Illumina HiSeq X-ten platform (Illumina,USA) using PE150 sequencing at Personal Biotechnology Co.,Ltd.(Shanghai,China).
2.3.Sequence analysisQuality-filtered reads were obtained from the raw sequencing reads for further analysis.First,sequencing adapters were removed from reads using Cutadapt(v.1.2.1) (Martin,2011).Low quality reads were then trimmed using a sliding-window algorithm.Third,reads that mapped to the host genome using the BWA (http://bio-bwa.sourceforge.net/) aligner (Li and Durbin,2009) were filtered out to remove host contamination.The quality-filtered reads were thende novoassembled to construct metagenomes for each sample using the iterative De Bruijn graph assembler for sequencing data with highly uneven depths (IDBA-UD) (Penget al.,2012).Coding regions (CDS) within metagenomic scaffolds of length >300 bp were predicted using MetaGeneMark (http://exon.gatech.edu/GeneMark/metagenome) (Zhuet al.,2010).The CDS from all samples were then clustered based on a 90% protein sequence identity threshold using CD-HIT (Fuet al.,2012) to obtain a non-redundant gene catalog.The gene abundance in each sample was then estimated with the SOAP program (http://soap.genomics.org.cn/) based on numbers of aligned reads.The lowest common taxonomic ancestors for non-redundant genes were obtained by comparing them against the National Center for Biotechnology Information (NCBI) non-redundant (nr)database by BLASTn (e value <0.001 used to identify matches).In addition,the functional profiles of non-redundant genes were obtained by annotating against the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Evolutionary Genealogy of Genes: Non-supervised Orthologous (EggNOG) databases individually using the DIAMOND (Buchfinket al.,2015)alignment algorithm.
2.4.Data accessAll raw sequence data were deposited in the NCBI Sequence Read Archive under accession numbers SAMN28013755–SAMN28013763.
2.5.Statistical analysesSpecies accumulation curves were used to assess the observed species richness of gut microbial communities with increasing sampling effort and ultimately to evaluate whether sufficient sampling effort was conducted.Accumulation curves were visualized in R based on the total number of species in each sample.Linear Discriminant Effect Size (LEfSe) analysis was conducted based on the taxonomic and functional profiles of non-redundant genes in order to detect taxa and functions with significant differences across the WL,NHB,and HB groups using the default parameters(Segataet al.,2011).Alpha diversity analysis was conducted to analyze the richness and uniformity of microbial community diversity across samples by calculating the Shannon diversity index using the Quantitative Insights Into Microbial Ecology(QIIME) software suite (Caporasoet al.,2010).To compare diversity among samples,species level composition was randomly resampled using the number of sequences in the smallest sample library in order to mitigate bias from differences in sequencing effort.Shannon diversity index values were calculated for each sample using the QIIME software program.Differences in Shannon diversity index between groups were evaluated with ANOVA tests using the R software environment.These data met assumptions of normality and equal variance.Pearson correlation analysis was conducted based on the Shannon diversity index when considering functional and taxonomic diversity using the R software environment.Differences in alpha diversity levels were evaluated based onRandPvalue correlation coefficients.Beta diversity analysis was also performed to investigate variation in microbial community composition and function across samples using the Bray-Curtis distance metric (Bray and Curtis,1957).Beta diversity analysis was conducted based on species compositional differences among samples.Beta diversity results were visualized with principal coordinates analysis (PCoA),nonmetric multidimensional scaling (NMDS),and unweighted pair-group method with arithmetic means(UPGMA) hierarchical clustering (Ramette,2007) using the QIIME program.PERMANOVA (Permutational multivariate analysis of variance) was used to compare the differences in beta diversity index between groups (Anderson,2001).Procrustes analysis (https://en.wikipedia.org/wiki/Procrustes_analysis) was used based on PcoA ordinations to assess whether microbial community functional and taxonomic variation were consistent with each other,as assessed withM2andPvalues in the QIIME program.The SciPy package for Python was used to compare the relative abundances of microbial phyla and genera among different groups usingttests.Pvalues were corrected in combination with the False Discovery Rate (FDR)(Benjamini and Hochberg,1995).Cladograms with circular representations of taxonomic hierarchies were generated using GraPhlAn (Asnicaret al.,2015).Histograms of KEGG metabolic pathway relative abundances in addition to histograms and violin plots of phylum-/genus-level taxonomic compositions were drawn using the Personal Genes Cloud Platform (https://www.genescloud.cn/chart).A heatmap showing KEGG metabolic pathway variation was also constructed using the Personal Genes Cloud Platform.
3.Results
3.1.Mangshan pit viper gut microbial taxonomic and functional compositionA total of 688,674,782 raw pairedend reads were generated using the Illumina HiSeq 2500 platform (Table S1).After filtering the sequence data,a total of 10,041,711,158 bp high quality sequence data were obtained(Table S2).De novoassembly led to the generation of at least 50,874 contig sequences and 49,269 scaffold sequences for each sample (Table S3).The metagenome length (total number of bases) obtained for each sample was at least 82,488,878 bp(contigs) and 82,275,992 bp (scaffolds) (Table S3).The assembled scaffolds exhibited N50 lengths ranging from 2,207 bp to 11,876 bp.A total of 1.05 million non-redundant genes were identified.Species accumulation curves for all samples indicated flattening of the curve(Figure 1A),suggesting that the metagenomic sequencing depth used here was sufficient to reflect actual microbial community richness within the samples.
TheP.mangshanensisgut microbial communities comprised four superkingdom level groups including bacteria that accounted for 93.68% of all sequence data,followed by eukaryotes (0.74%),viruses (0.36%),and archaea (0.02%),with the remaining 5.21% comprising unclassified sequence data.A total of 4,408 microbial species were identified that were affiliated with 64 phyla,163 classes,332 orders,634 families,and 1,576 genera (including unknown taxa).The most abundant phyla were Proteobacteria (65.55%),Bacteroidetes (15.97%),and Firmicutes (8.11%) (Figure 1B).Approximately 0.1% of the eukaryotic reads comprised three phyla including Ascomycota,Apicomplexa,and Basidiomycota (Figure 1C).The most abundant archaeal phylum was Euryarchaeota (0.02%) (Figure 1D).The five most abundant genera wereEscherichia(22.34%relative abundance),Bacteroides(13.85%),Salmonella(6.47%),Morganella(6%),andPseudomonas(5.6%) (Figure S1).
Variation in protein abundances were obtained based on KEGG annotations of non-redundant protein sequences and a protein abundance matrix.A total of 440,136 KEGG pathways were identified among all samples including 241,638 that corresponded to “metabolism”,42,575 that corresponded to “environmental information processing”,and 41,261 that corresponding to “genetic information processing”,among others (Figures 1E and S2).The most prevalent sub-category within metabolic pathways (metabolic pathway subfunctions)included carbohydrate metabolism (68,143 pathways),amino acid metabolism (43,752),and membrane transport(26,304) (Figures 1F and S2).Similar results were obtained using eggNOG database annotations,wherein the microbial communities were enriched in “metabolism”,“cellular processes and signaling”,and “information storage and processing”,among other functions (Figure S3;Table S4).
3.2.Captivity and hibernation alter the diversity and composition of gut microbial communitiesWild Mangshan pit vipers exhibited higher microbial taxonomic diversity (based on Shannon indices) than non-hibernating Mangshan pit vipers (F=32.55,P=0.005),whereas diversity differences between wild and hibernatingP.mangshanensis(F=1.45,P=0.29),in addition to hibernating compared to nonhibernating Mangshan pit vipers (F=3.52,P=0.134),were not significant (Figure 2A).PcoA (Figure 2B;PERMANOVA,F=7.31,P=0.003) and NMDS (Figure 2C;PERMANOVA,F=7.31,P=0.003) ordinations of Bray-Curtis distances among the three groups indicated that the microbial communities were significantly separated by group.Inter-group Bray-Curtis distances were markedly higher than intra-group distances,indicating that the species composition of samples from the same group were highly similar.Similar results were observed for UPGMA cluster analysis (Figure 2D).Moreover,cluster analysis indicated that microbial communities were more similar between wild snakes and hibernating snakes,but those from non-hibernating snakes were quite different from the other two groups (Figure 2D).
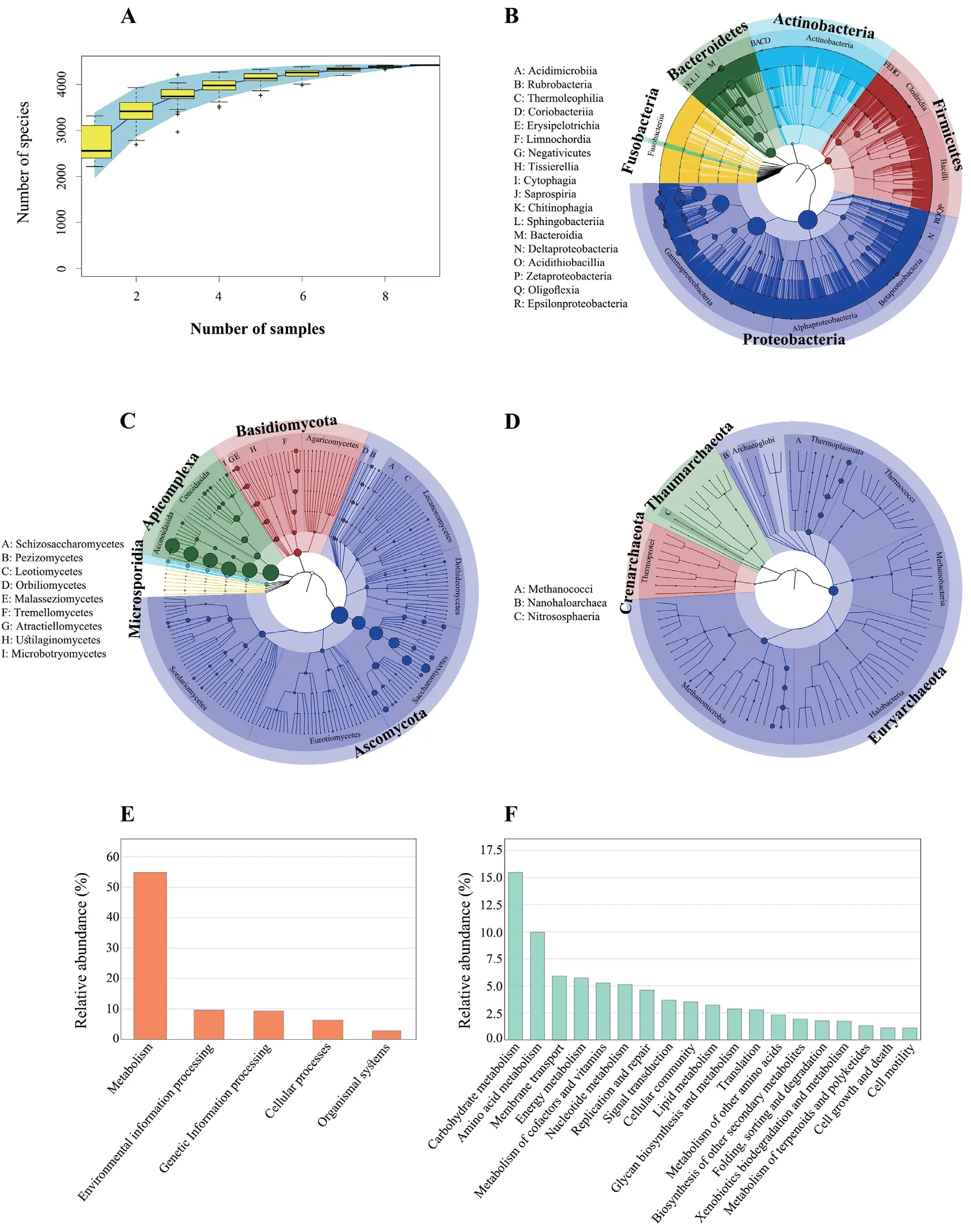
Figure 1 Taxonomic and functional profiling of Mangshan pit viper gut microbial communities.(A): Species accumulation curve of the taxonomic diversity within P.mangshanensis gut microbial communities.The x axis represents sampling effort,and the y axis represents the number of detected microbial species.Phylogenetic trees showing the (B) bacterial,(C) eukaryal,and (D) archaeal taxonomic composition withing P.mangshanensis gut microbial communities.Extending from the inner circle to the outer circle,the classification tree shows the hierarchical relationships at all taxonomic levels (represented by nodes) from the domain to species level.The node size corresponds to the average relative abundance of each taxonomic group,and different colors indicate different taxonomic groups.The relative abundances of proteins in the first and second tier metabolic pathways of KEGG are shown in (E) and (F),respectively.Proteins related to human diseases or without functional annotation were excluded from the analysis.In addition,only secondary metabolic pathways with relative abundance >1% are shown.
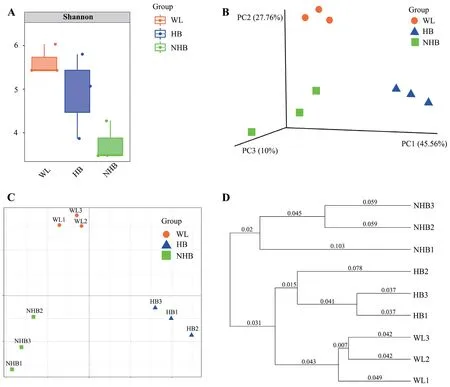
Figure 2 Comparison of Mangshan pit viper gut microbial community diversity across groups.(A): Differences in Shannon diversity index values among experimental groups.The Shannon index considers community richness and evenness,wherein higher values indicate greater community diversity.(B): Three-dimensional PCoA analysis of among-community differences based on Bray Curtis distances.More similar communities are more closely arranged in ordination space.The percentages in brackets indicate the variation in among-community distances that can be interpreted by the corresponding axis.(C): Two-dimensional non-metric multidimensional scaling (NMDS) analysis of among-community differences based on Bray Curtis distances.More similar communities are more closely arranged in ordination space.(D): UPGMA clustering of samples based on Bray Curtis distances,which clusters samples according to their dissimilarities.Shorter branch lengths indicate more similar samples.WL: wild P.mangshanensis group,NHB: non-hibernating P.mangshanensis group,HB: hibernating P.mangshanensis group.
The relative abundance profiles at the phylum and genus levels indicated distinct microbial community characteristics among groups (Figure 3A–B).Proteobacteria,Firmicutes,Bacteroidetes,and Actinobacteria were the dominant phyla among the three groups (mean relative abundance >1%).However,Firmicutes proportions were significantly higher in wildP.mangshanensisthan in hibernatingP.mangshanensis(P=0.0004,FDR=0.008) and non-hibernatingP.mangshanensis(P=0.0003,FDR=0.008).The relative abundances of the generaBlautia,Clostridium,andLachnoclostridium(Figure 4A–C) were significantly higher in the WL group communities than in the HB group (P=0.002,FDR=0.047;P=0.01,FDR=0.129;P=0.01,FDR=0.117),and in the NHB group (P=0.004,FDR=0.082;P=0.044,FDR=0.197;P=0.009,FDR=0.102),which all represent genera that can produce short-chain fatty acids (SCFA).The relative abundances of opportunistic pathogens includingAeromonas,Klebsiella,andSalmonella(Figure S4) that can cause enteritis were also higher in the WL group communities compared to the other two groups.Several anaerobic genera includingStenotrophomonas,Comamonas,Alcaligenes,andBordetella(Figure S5),were the most abundant microbial genera in the HB group and all of them belong to the Proteobacteria phylum.The relative abundances ofAkkermansiawere also significantly higher in the HB group than in the WL group(Figure 4D;P=0.02,FDR=0.151),and in the NHB group (Figure 4D;P=0.023,FDR=0.161).In addition,the relative abundances ofBifidobacteriumandLactobacilluswere higher in the NHB group than in the other two groups (Figure 4E–F).It is worth noting that more opportunistic pathogens were observed in the NHB group communities includingFusobacterium,Muribaculum,Shigella,Escherichia,andMorganella(Figure S4).In addition,the relative abundances of carbohydrate metabolism and protein metabolism-related microbial taxa (e.g.,Adlercreutzia,EubacteriumandParabacteroides)were higher in the NHB group compared to the other two groups (Figure 4G–I).Among them,the relative abundance ofAdlercreutziaandParabacteroidesin the NHB group was significantly higher than that in the WL group (Figure 4G,P=0.041,FDR=0.28;Figure 4I,P=0.035,FDR=0.263),and the relative abundance ofEubacteriumandParabacteroidesin the NHB group was significantly higher than that in the HB group (Figure 4H,P=0.033,FDR=0.252;Figure 4I,P=0.042,FDR=0.283).
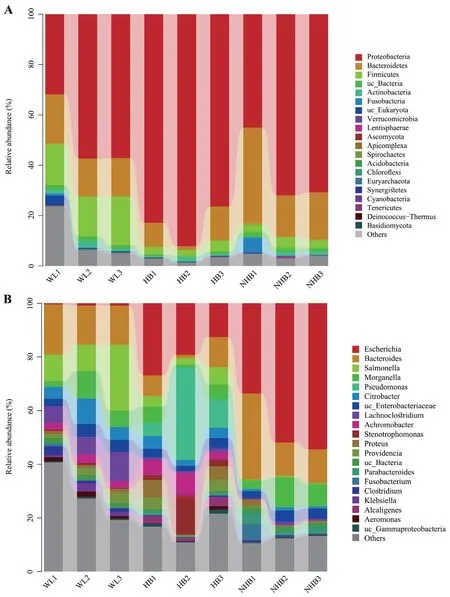
Figure 3 Relative abundances of taxonomic groups among P.mangshanensis gut community samples at the (A) phylum and (B) genus levels.Cumulative taxonomic abundances are shown as bar charts.Only the 20 most abundant phyla or genera are shown.
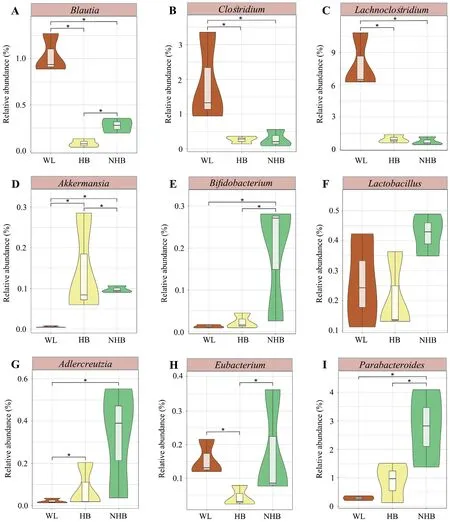
Figure 4 Relative abundance comparisons of P.mangshanensis gut microbial community genera that exhibit important functions.(A)-(I) show genera with important functions that exhibited significantly different relative abundances between groups.Different groups in the violin plots are represented by different colors.Significance of differences between groups was evaluated with t tests,and FDRs were corrected using a Benjamini-Hochberg correction.Asterisks (*) indicate statistical significance at P <0.05 between groups.
To assess differences in microbial communities affected by captivity and hibernation differences,the cladogram generated by LefSe analysis was used to screen the differential microbes among groups (Figure S6).The gut microbiota composition of wild snakes differed from those in captive snakes including several Firmicutes and Proteobacteria taxa (e.g.,Aeromonas,Klebsiella,andSalmonella) (Figure S4).In addition,hibernating and non-hibernating captive conditions corresponded to major shifts in microbiota composition,with higher relative abundance in several Proteobacteria and Bacteroidetes taxa (e.g.,Fusobacterium,Muribaculum,Shigella,Escherichia,andMorganella)in non-hibernating Mangshan pit vipers (Figure S4) along with several proteobacterial taxa in hibernating Mangshan pit viper communities (e.g.,Alcaligenes,Bordetella,Comamonas,andStenotrophomonas) (Figure S5).
3.3.Effects of captivity and hibernation on gut microbiome functionsPearson correlation analysis of functional and taxonomic diversity based on alpha diversity analysis (assessed with the Shannon index) indicated inconsistent diversity across groups when comparing the two community characteristics(Figure 5A;R=0.44,P=0.24).However,beta diversity analyses revealed consistent patterns at the functional and taxonomic levels (Figure 5B;Monte Carlo permutation test,M2=0.16,P<0.001).
Comparison of KEGG functional profiles among groups indicated that wildP.mangshanensismicrobiota were enriched in secondary pathways including membrane transport,signal transduction,signaling molecules and interactions,nucleotide metabolism,and immune system pathways (Figure 5C).A greater diversity in secondary pathways were enriched in hibernating Mangshan pit viper microbial communities including cell motility,cellular community,signal transduction,amino acid metabolism,the metabolism of terpenoids and polyketides,xenobiotic biodegradation and metabolism,aging,circulatory system,and environmental adaptation pathways,among others (Figure 5C).In addition,non-hibernatingP.mangshanensisalso exhibited unique enrichment in secondary pathways including cell growth and death,replication and repair,carbohydrate metabolism,lipid metabolism,and glycan biosynthesis and metabolism (Figure 5C).
Furthermore,the relative abundance of several thirdlevel pathways was higher in wild Mangshan pit viper gut communities including ABC transporters and phosphotransferase systems (PTS) within membrane transport,and the biosynthesis pathways of ansamycins within the metabolism of terpenoids and polyketides (Figure 6A–B).In contrast,several third-level pathways were higher in hibernating Mangshan pit viper communities including those involved in sulfur metabolism,bacterial chemotaxis,and phenylalanine metabolism (Figure 6A and 6C).In addition,the HB group gut communities exhibited significantly higher abundances of ABC transporters (P<0.001,FDR=0.002) and two-component systems (P=0.011,FDR=0.023) compared to the NHB group communities (Figure 6B).In non-hibernatingP.mangshanensismicrobial communities,other glycan degradation metabolic pathways in the broader glycan biosynthesis and metabolism category,along with sphingolipid metabolism within the lipid metabolism category,were higher compared to communities in the other two groups (Figure 6B–C).Lastly,the pentose phosphate pathway (P=0.002,FDR=0.036),cysteine and methionine metabolism (P<0.001,FDR=0.002),and galactose metabolism (P<0.001,FDR=0.003) pathway abundances were significantly higher in the NHB group gut communities compared to the HB communities (Figure 6C).
4.Discussion
Increasing numbers of studies of gut microbiota in vertebrates have been reported in recent years,coinciding with the rapid development of high-throughput sequencing (next-generation sequencing) technologies (Leyet al.,2008a;Leyet al.,2008b;Sullamet al.,2012;Waite and Taylor,2014;Kohlet al.,2017;Huanget al.,2018a;Tonget al.,2020;Levinet al.,2021).However,our understanding of reptilian gut microbiomes is still relatively minimal compared to birds or mammals,despite that they are critical organisms for understanding the evolution of vertebrates (Jianget al.,2017;Kohlet al.,2017;Zhanget al.,2019;Tonget al.,2020).Further,many squamate reptile species are facing greater global loss crises in recent years,based on IUCN classifications.Thus,the study of microbiota within endangered reptile species could provide a scientific basis for additional conservation efforts including artificial breeding,disease prevention,and reintroduction (Colston,2017).
4.1.Dominant gut microbial community members withinP.mangshanensisLike most reptiles,the dominant gut microbial taxa within Mangshan pit vipers were Proteobacteria,Bacteroidetes,and Firmicutes.The dominant bacterial phyla of theP.mangshanensisgut communities were similar to those identified inAgkistrodon piscivorus,four farmed snakes from southern China (Naja atra,Ptyas mucosa,Elaphe carinata,andDeinagkistrodon acutus),and three species of venomous Asian snakes (Laticauda laticaudata,Trimeresurus flavomaculatus,andBoiga dendrophila) (Colstonet al.,2015;Zhanget al.,2019;Smithet al.,2021).However,there were differences in the relative abundances of dominant bacterial phyla in theP.mangshanensisgut microbiomes compared to the above snake species (Table S5).Moreover,the dominant bacterial phyla of the Mangshan pit viper gut communities differed from those identified in the gut microbiota of Burmese pythons (Python bivittatus) (i.e.,dominated by Firmicutes and Bacteroidetes)(Costelloet al.,2010),timber rattlesnakes (Crotalus horridus)(i.e.,uniquely dominated by Proteobacteria) (McLaughlinet al.,2015),or Northern Watersnakes (Nerodia s.sipedon) (i.e.,uniquely dominated by Tenericutes,Proteobacteria,Firmicutes,and Bacteroidetes) (Dallaset al.,2021).Thus,the composition and relative abundances of the dominant bacterial phyla of theP.mangshanensisgut microbiota exhibited both unique characteristics as well as shared characteristics relative to communities from other snakes.Nevertheless,it should be noted that it is difficult to compare 16S rRNA gene sequencing results from different studies,as sampling and sequencing methods could influence overall results.Few overall studies have investigated the gut microbial communities of snakes.Thus,the composition and function of eukaryotic and archaeal populations in gut microbiota are not fully understood (Zhanget al.,2019;Dallaset al.,2021;Smithet al.,2021).Although these groups account for a relatively small percentage of the entire gut microbiomes,they may play important and specific roles in the metabolic,immune,or other functions ofP.mangshanensis(Eckburget al.,2005;Handlet al.,2011;Fosteret al.,2013;Yanget al.,2018).The detection and quantification of viruses are challenging due to the high genetic diversity within viral genomes,the diversity of genes carried by viruses,and the lack of conserved genetic elements among viruses (Swansonet al.,2011).
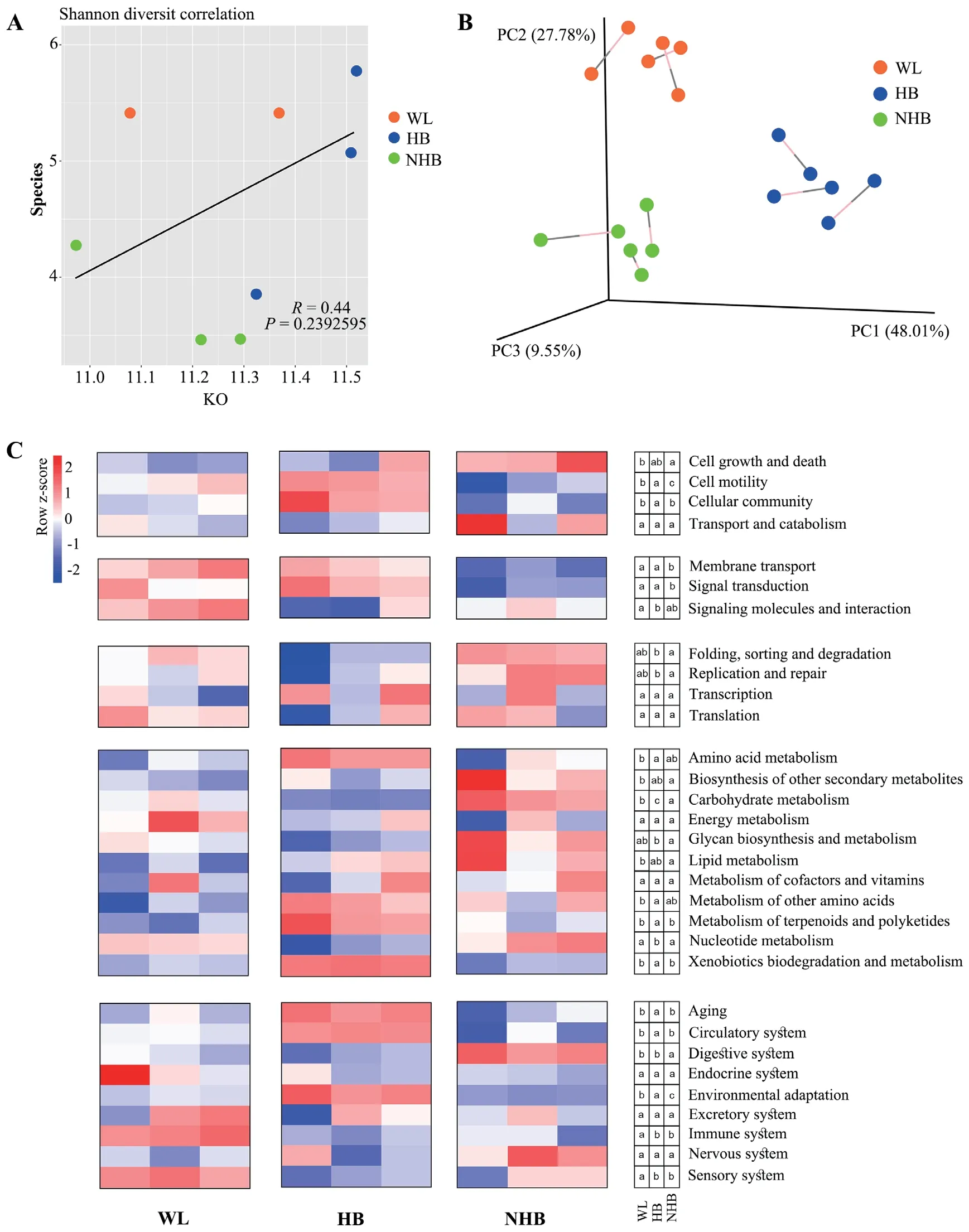
Figure 5 Comparison of gut microbiota functions across different Mangshan pit viper groups.(A): Pearson correlation analysis of the Shannon diversity index at the taxonomic and functional levels of gut communities.(B): Procrustes analysis of PCoA ordinations of taxonomic and functional variation among samples.The percentage in brackets indicates the variation in among-community distances that can be explained by each axis.(C): Heatmap showing the abundances of KEGG secondary functional pathways of gut microbial communities among wild Mangshan pit viper (WL group),non-hibernating Mangshan pit viper (NHB),and hibernating Mangshan pit viper (HB).The relative abundance of each pathway is colored according to the row z-score ((value– row mean)/row standard deviation).The lowercase letters in the three small boxes in the same row on the right side of the heatmap represent the difference in the relative abundance of a certain pathway (shown on the far right) between the WL group,the HB group and the NHB group.The groups that did not share lower case letters were statistically significant from one another according to the one-way ANOVA with the LSD post hoc test.The letter “a” indicates the highest relative abundance for the pathway in that group,followed by the letter “b” and “c” for lesser relative abundances.
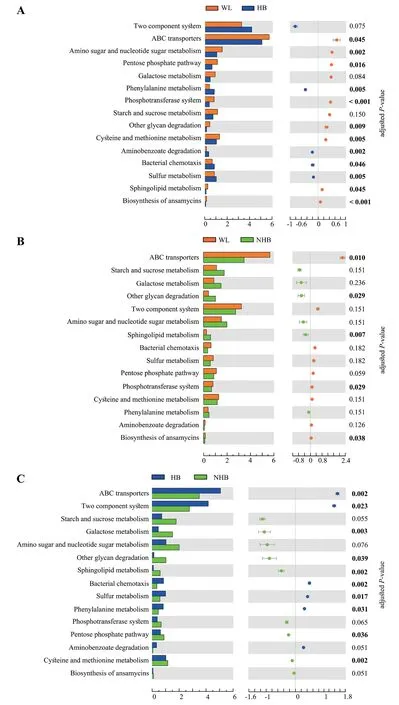
Figure 6 Comparison of relative abundances of third-level KEGG functional pathways among Mangshan pit viper gut microbial communities.(A)-(C): Differences in third-level functional pathway relative abundances pathways between groups.Statistical significance was assessed using t tests and P values were adjusted by the FDR.Numbers in bold denote a significant difference (adjusted P value <0.05).
4.2.Effects of captivity and hibernation on P.mangshanensis gut microbiotaThe gut microbial composition of wild Mangshan pit vipers differed from those of captive Mangshan pit vipers,with gut microbiota diversity of wild Mangshan pit vipers also being higher than in non-hibernating Mangshan pit viper microbiota.The gut microbial community diversities in free-range animals are often higher than in captive animals because wild animals are exposed to natural environments and thus,more diverse microbial communities that then shape their gut microbial communities (Huanget al.,2018b).Moreover,among the diverse environmental influences on gut microbial communities in the wild,dietary diversity represents one of the most important factors responsible for driving diversity of gut microbial communities in wild individuals (Sandriet al.,2020).The gut microbial communities within the same group ofP.mangshanensiswere most similar,indicating that individuals with the same living conditions acquired gut microbiome in similar manners (Figure 2D).This is consistent with typically observed differences in gut community composition between captive and wild populations of animals (Renet al.,2016;Tanget al.,2020;Eliadeset al.,2021).Previous studies have shown that captivity alters gut microbiota abundances,although host-specific communities are nevertheless retained (Kohlet al.,2017).The results of this study are consistent with these previous observations.The dominant gut microbial taxa within Mangshan pit vipers (i.e.,the phyla Proteobacteria,Firmicutes,Bacteroidetes,and Actinobacteria,in addition to the generaEscherichia,Bacteroides,Salmonella,Morganella,andPseudomonas) were the same despite different living environments (Figures 1B and 3,Figure S1).Thus,these taxa are likely to derive from vertically transmitted core microbiota that may be important symbionts that participate in host nutrition,metabolism,and immunity.
As previously reported,mucosal microbiota may survive during host hibernation via host-derived mucin glycan (Tanget al.,2019).It is worth noting that mucin degradation may be a pathogenic process since it destroys the mucosal barrier,leads to intestinal inflammation,increases the permeability of the intestinal mucosa,and exposes gastrointestinal cells to harmful substances,consequently resulting in the invasion of opportunistic intestinal pathogens (Thomaset al.,2011).However,some studies have shown that the expression of caecal receptor proteins (e.g.,MUC2,TLR4,and TLR5) was differentially regulated during animal hibernation,suggesting the presence of a protective response that minimizes inflammation (Dill-Mcfarlandet al.,2014).In addition,the intestinal immune system of ground squirrels has been observed to be remodeled during hibernation to promote tolerance of microbiota and suppress inflammatory pathways (Kurtz and Carey,2007;Dill-Mcfarlandet al.,2014).Studies of Chinese alligator gut microbiomes have also shown the presence of highly expressed paralogous β-defensins in gastrointestinal tracts during hibernation that help them fend off opportunistic pathogens,and which might help them maintain intestinal epithelial integrity and function during winter fasting (Tanget al.,2019).Therefore,we suggest that rearing Mangshan pit vipers with artificial hibernation should be considered an important management strategy for this endangered snake,because it may help snakes stimulate their intestinal immune systems and maintain intestinal homeostasis.
Gut microbial taxa are generally defined as either ‘beneficial’or ‘harmful’ based on host-microbe interactions (Cirsteaet al.,2018).Beneficial microorganisms can be used in microbialbased interventions to regulate gut homeostasis and promote host health (Kunduet al.,2017;Cirsteaet al.,2018).In this study,the beneficial microbial taxa differed within the gut microbial communities of the three groups of snakes.The gut microbiota of wild snakes was enriched in beneficial strains that produce SCFAs includingBlautia,Clostridium,andLachnoclostridium(Figure 4).SCFAs,and especially butyrate,provide up to 70%of the energy required by colonic epithelial cells and thus play important roles in maintaining colonic mucosa homeostasis(Roediger,1980;Huanget al.,2018b).Akkermansiaexhibited higher relative abundances in hibernatingP.mangshanensisand may be involved in gut remodeling and autophagy as a component of gut homeostasis (Figure 4D) (Kunduet al.,2017;Cirsteaet al.,2018).
Non-hibernatingP.mangshanensisare fed throughout the year,potentially resulting in functional specialization of their gut microbial communities,which are enriched in organisms associated with carbohydrate and lipid metabolism.Although several beneficial microbial taxa were detected in non-hibernatingP.mangshanensis(e.g.,BifidobacteriumandLactobacillus) (Figure 4),more pathogenic microorganisms were evident in their gut microbial communities.In particular,the gut microbiota of non-hibernating Mangshan pit vipers was dominated byEscherichia coliat the species level (Figure S7).E.colihas historically been considered extremely virulent pathogens that can cause acute hemorrhagic diarrhea and inflammation of hibernatingP.mangshanensisintestinal mucosa (Yantisset al.,2007;Cirsteaet al.,2018).Several other pathogenic taxa were also identified in the microbiota of the other two groups,includingEscherichiaandSalmonellain wildP.mangshanensis.Previous studies in animals and humans have suggested that gut inflammation produces nitrates that stimulateE.coliandSalmonellagrowth (Lopezet al.,2012;Winteret al.,2013).Other studies have shown that continuous contact with breeders and feed buildings provide increased opportunities for transmission of pathogenic microbiota from host-associated and environmental sources that can colonize the guts of captive animals (Nelsonet al.,2012;Tanget al.,2020).The gut microbiota profiles of captive animals can change due to increasing anthropogenic pressures,potentially leading to increased pathogen and viral titers compared with hibernating animals (Tanget al.,2019).Keeping snakes captive without hibernation can make snakes gain additional weight,while the results of this study indicate that more opportunistically pathogenic bacteria were present in the gut microbiomes of non-hibernating Mangshan pit vipers.As mentioned above,continuous contact with breeders and feed buildings provides increased opportunities for transmission of potentially pathogenic microorganisms (Nelsonet al.,2012;Tanget al.,2020).Consequently,the above reasons may underlie the increased risk of gut diseases and ultimately limit the growth of non-hibernatingP.mangshanensispopulations.
4.3.Functional specialization of Mangshan pit viper gut microbiomesHibernation reorganizes the host’s gut microbiota and may also stimulate the intestinal immune system,while animals often need to improve food digestibility and nutrient transport during their active period to store enough fat for hibernation (Dill-Mcfarlandet al.,2014;Tanget al.,2019).Interestingly,we observed that individuals in the WL and HB groups harbored gut communities with significantly improved membrane transport functions,and especially ABC transporter functions,relative to those in NHB individuals(Figures 5C and 6).Previous studies have observed that numerous substrates are transported by ABC transporters,including various small and macromolecular compounds like carbohydrates,amino acids,vitamins,polypeptides,proteins,and cell metabolites,among others (Higgins,1992).ABC transporters also participate in many important physiological processes including nutrient uptake,cell detoxification,abnormal lipid homeostasis,and non-classical secretion of signaling molecules and toxins,among others (Schmitt and Tampé,2002;Panet al.,2016).The results of this study also provide confirmatory evidence for identifying whether animals that hibernate regularly harbor gut microbiomes that are active in nutrient transport,absorption,and intestinal immune regulation.Among phosphotransferase systems (PTS) and biosynthesis pathways of ansamycins,wild Mangshan pit viper communities were also higher in their abundances compared to captive individuals (Figure 6C).Phosphate is one of the most abundant mineral compounds in animals.Phosphate homeostasis in bodies is primarily controlled by intestinal absorption,retention or release by bones,and reabsorption via kidneys.These dynamics are necessary for many biological processes,including skeletal development and bone integrity,energy metabolism,cell signaling,and the regulation of protein synthesis (Markset al.,2010;Sabbaghet al.,2011).The biosynthesis of ansamycins is related to the production of antibiotics that could inhibit potential pathogens in host intestines (Songet al.,2021b).Therefore,the gut microbiota of wild Mangshan pit vipers is conducive to nutrient metabolism and transportation,signal transduction,and immune regulation,which may be particularly important for gut homeostasis and overall health of snakes.
The population of artificially hibernating snakes analyzed in this study was relatively stable.The gut communities of this group of snakes exhibited several significantly enriched pathways compared to those in the other two groups,including pathways involved in bacterial chemotaxis,sulfur metabolism,and phenylalanine metabolism (Figure 6).Among them,chemotaxis is a sensory system within bacteria that is used to regulate and respond to external environmental stimuli;it is the most well-known model system for bacterial signal transduction (Eisenbach,1996).Bacteria use complex and effective chemotaxis systems to identify favorable conditions and increase their adaptations to environments (Tindallet al.,2012;Yanget al.,2015).In addition,sulfur is an essential chemical element for all organisms and is used as a structural component of biomolecules,but also plays a specific role in metabolism (Carboneroet al.,2012).Lastly,aromatic amino acids,including phenylalanine,tyrosine,and tryptophan,can be used to produce important bioactive substances via metabolism and are also essential dietary amino acids for animals (Bender,2012).The enrichment of pathways related to the above areas in hibernating snakes may help regulate the adaptation of gut microbiomes to complex environments,which may underscore the need to maintain gut homeostasis by maintaining gut microbiota stability.
In contrast to wild snakes and snakes that hibernate,the function of gut microbial communities in non-hibernatingP.mangshanensisthat do not hibernate warrant additional research attention.This study demonstrated that metabolic and digestive system-associated physiological pathways were significantly enriched in the gut microbiota of non-hibernating Mangshan pit vipers,and especially pathways involved in lipid and carbohydrate metabolism (Figure 5C).Moreover,significantly increased abundances of KEGG third-level pathways including other glycan degradation and sphingolipid metabolism pathways in non-hibernating vipers compared with the other two snake groups also suggest that gut microbial communities in the former group primarily function in metabolizing nutrient substrates within the intestine (Figure 6B–C).Snakes are carnivores and their primary food sources are rich in protein,fat,and vitamins,but only contain small amounts of carbohydrates (Costelloet al.,2010).However,carbohydrate metabolism pathways were significantly enriched among the gut microbial communities of non-hibernatingP.mangshanensisin this study (Figure 5C).Captive Mangshan pit vipers are fed small white mice,and thus carbohydrates metabolized byP.mangshanensisgut microbiota only likely derive from the gut contents of mice.Carnivores have short digestive tracts,generally lack digestive enzymes,and have a high demand for amino acids (Leyet al.,2008b;Costelloet al.,2010;Choet al.,2013;Sousa-Pereiraet al.,2015).Indeed,excessive intake of carbohydrates can burden their metabolism (Costelloet al.,2010;Kimet al.,2016).In addition,eating has substantial benefits in terms of both nutrient and energy acquisition for snakes and other reptiles,although the digestion and metabolism of food also incurs negative physiological costs and increases oxidative damage of cells or tissues (Butleret al.,2016).Non-hibernating,captiveP.mangshanensiswere active year-round in this study.Containment in a feeding space for a long time and constant interaction with breeders may promote the accumulation of potentially pathogenic microorganisms in intestines (Eliadeset al.,2021).Further,continuous eating can alter gut microbial community functional enrichment and specialization within captive snakes,thereby resulting in intestinal oxidative damage (Butleret al.,2016;McKenneyet al.,2018).In conclusion,feeding Mangshan pit vipers without hibernation may lead to damaged intestinal homeostasis that could be one of the reasons explaining their reduced populations.
Comparison of the composition,diversity,and function of gut microbial communities in Mangshan pit vipers under artificial hibernation and non-hibernating rearing conditions will help provide a scientific basis for improving the artificial rearing of this critically endangered species and contribute to its survival and health under extreme physiological conditions.Despite the limited sample size of this study,the results shown here nevertheless provide a preliminary framework for larger future studies.
5.Conclusions
The composition and function ofP.mangshanensisgut microbiota differed from those identified in other snakes and vertebrates.Specifically,the gut microbial communities of Mangshan pit vipers comprised 64 phyla,163 classes,332 orders,634 families,1,576 genera,and 4,408 species.The most abundant phyla in theP.mangshanensisgut microbiomes were the Proteobacteria,Bacteroidetes,and Firmicutes.KEGG pathway analysis indicated that metabolism,environmental information processing,and genetic information processing were particularly enriched functions within the gut microbiota populations.Further,wild Mangshan pit viper gut microbial communities exhibited higher microbial taxonomic diversity than did non-hibernating Mangshan pit viper communities.In addition,the relative abundances of the dominant phyla and genera within the gut communities were distinct among wild,hibernating captive,and non-hibernating captive Mangshan pit vipers.Importantly,greater numbers of potential pathogens were enriched in the gut microbiota of non-hibernatingP.mangshanensis.Therefore,captivity and hibernation may deleteriously affect the diversity and composition of Mangshan pit viper gut microbial communities.Further,captivity and artificial hibernation led to differences in the functional pathways encoded by the gut microbiota populations.Among these differences,increased enrichment of metabolic pathways was encoded by the gut microbial communities of nonhibernating captive Mangshan pit vipers.In contrast,the gut microbiomes of wild and hibernating captive snakes may better reflect healthy intestinal homeostasis than those in nonhibernating captive snakes.Thus,our study suggests that the composition,diversity,and functions ofP.mangshanensisgut microbiota were altered,perhaps negatively,by different living environments and captivity methods.However,it should be noted that the small sample size of this study may limit its interpretation across broader comparisons.Nevertheless,the preliminary results of this investigation of Mangshan pit viper gut microbiomes provide a useful framework for future studies.
AcknowledgementsThis work was supported by the National Natural Science Foundation of China (Grant No.31472021),the Project for Wildlife Conservation and Management of the National Forestry and Grassland Administration of China (Grant No.2021-HN-001),and the Wildlife Conservation Project of Hunan Province (Grant No.HNYB2019-001).We are grateful to Jundong DENG,Xinguo GONG,and Qingsong JIANG for assistance in collecting samples.We thank Shuheng LI from the Qilu Normal University for providing help with referencing and manuscript preparation.
Appendix
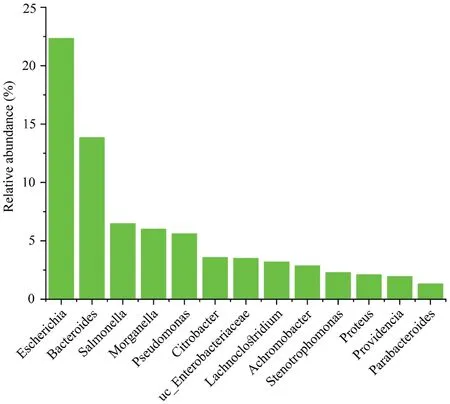
Figure S1 The most abundant genera (>1% relative abundance) in Mangshan pit viper.
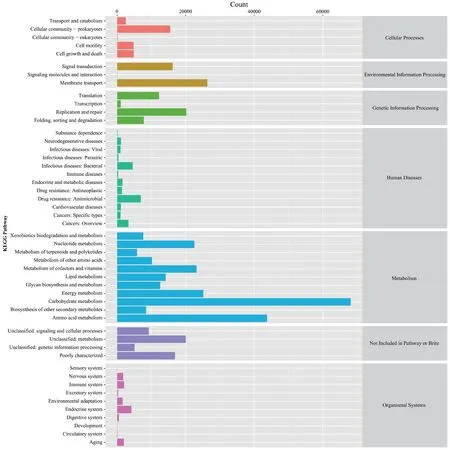
Figure S2 KEGG annotations of primary and secondary metabolic pathways within gut microbial communities.The number of proteins annotated to the corresponding metabolic pathway is shown on the x-axis and each secondary metabolic pathway is shown on the y-axis,while the first level classification of each metabolic pathway is shown on the right.
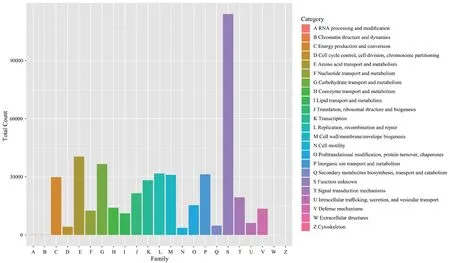
Figure S3 EggNOG database annotations of functions within gut microbial communities.The x-axis shows the 23 gene function categories of EggNOG,represented by different capital letters,and the y-axis shows the number of EggNOG functional groups annotated to the corresponding classification group.
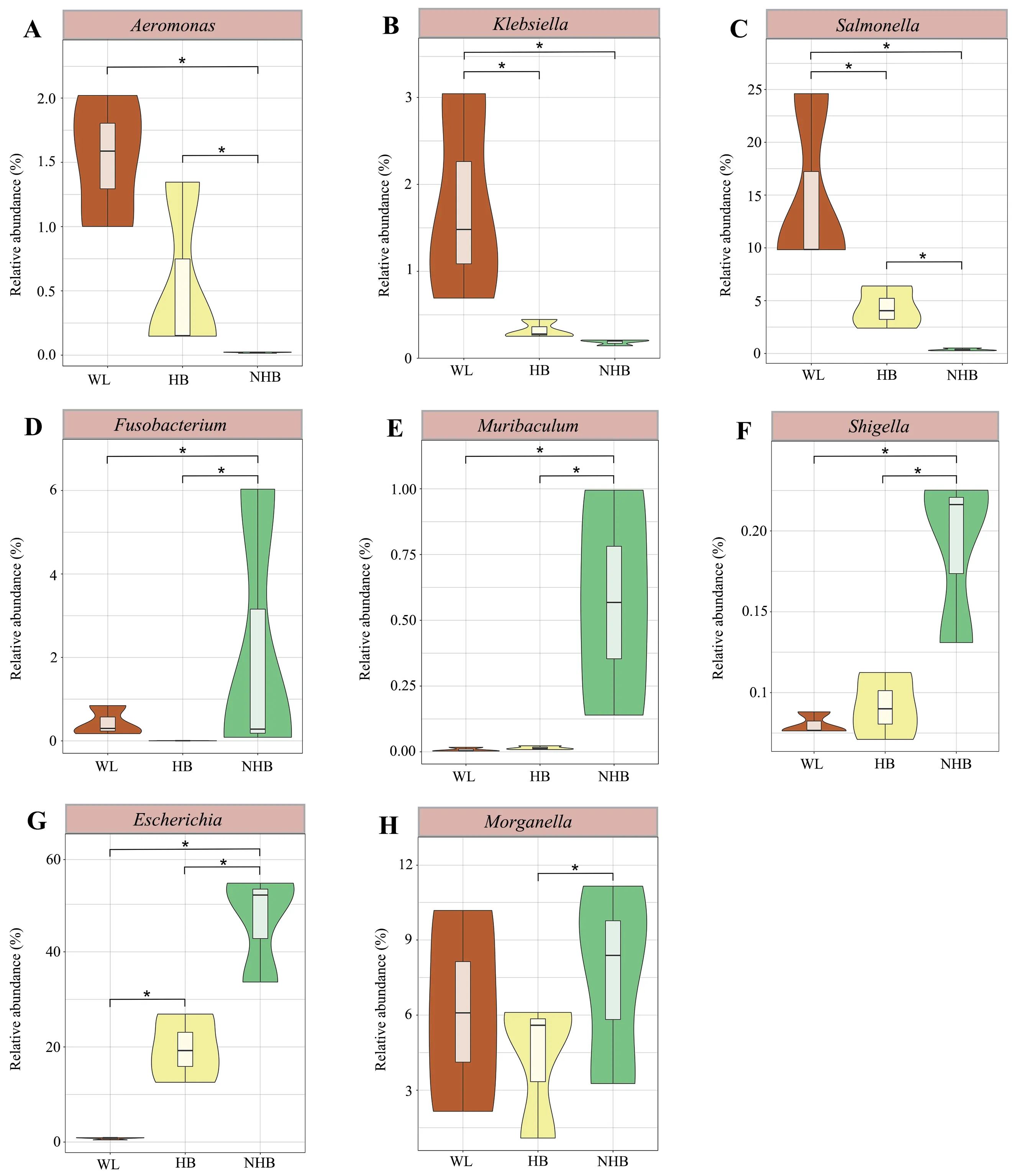
Figure S4 Violin plots showing the relative abundances of opportunistic pathogens among groups.Different groups are represented by different colors.The significance of differences between groups was evaluated by t test,and FDRs were corrected with Benjamini-Hochberg corrections.
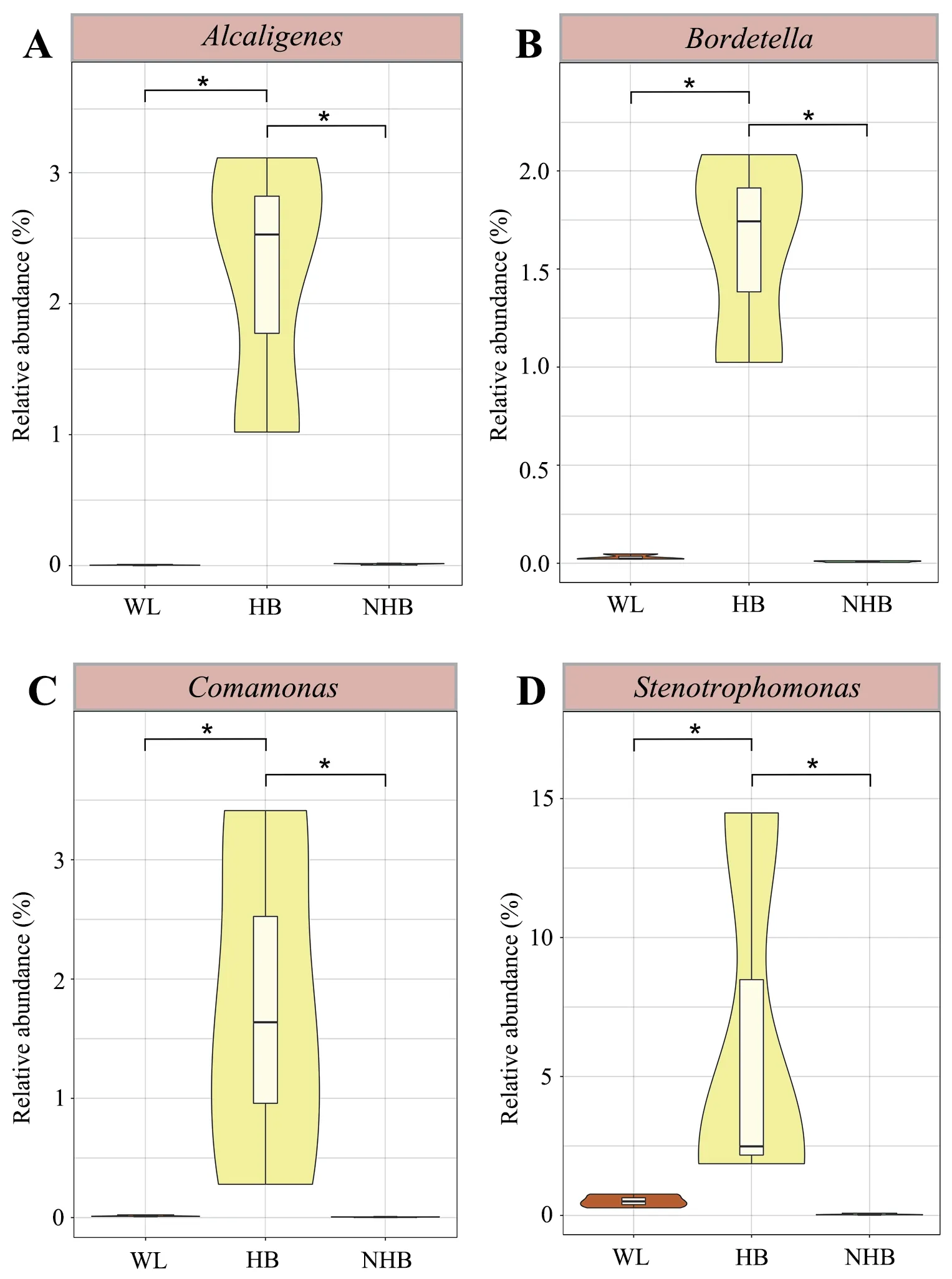
Figure S5 Violin plots showing the relative abundances of anaerobic bacteria among groups.Different groups are represented by different colors.The significance of differences between groups was evaluated by t test,and FDRs were corrected with Benjamini-Hochberg corrections.
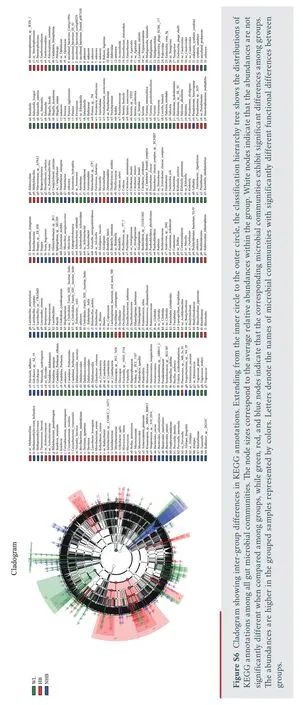
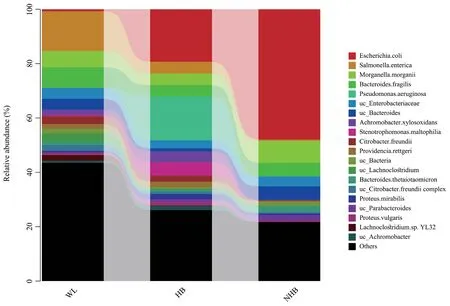
Figure S7 The relative abundance (%) of gut microbial communities from wild,hibernating,and non-hibernating Mangshan pit viper at species level.Cumulative taxonomic abundances are shown as bar charts.Only the 20 most abundant phyla or genera are shown.
The Tables S1–S5 can be downloaded from the website https://pan.baidu.com/s/1nfIJweRz54huVEV8HUJbYw (access code: bp7j).
杂志排行

Asian Herpetological Research的其它文章
- Three New Species of Diploderma Hallowell,1861 (Reptilia: Squamata:Agamidae) from the Shaluli Mountains in Western Sichuan,China
- Application of eDNA Metabarcoding for Detecting Anura in North China
- Phylogenetic Relationships among Chinese Rice Frogs within the Fejervarya limnocharis Species Complex (Amphibia: Dicroglossidae)
- Lineage Diversification and Niche Evolution in the Chinese Cobra Naja atra(Elapidae)
- Endocast Morphological Variation and Its Driving Forces in Scutiger boulengeri
- Desertification Drives the Shift in Egg Size-Number Trade-Off in an Agamid Lizard