Application of eDNA Metabarcoding for Detecting Anura in North China
2023-01-05WenhaoLIMingshuoQINXiangleiHOUJiaqiZHANGSiqiWANGYuLIZexuLUOTengDENGTianjianSONGChunxiaXUXuanLIUXuyuWANGandYimingLI1
Wenhao LI ,Mingshuo QIN,3 ,Xianglei HOU,3 ,Jiaqi ZHANG,3 ,Siqi WANG,3 ,Yu LI,3 ,Zexu LUO,3,Teng DENG,3,Tianjian SONG,3,Chunxia XU,3,Xuan LIU,3,Xuyu WANG and Yiming LI1,,3*
1 School of Life Sciences,Institute of Life Sciences and Green Development,Hebei University,Baoding 071002,Hebei,China
2 Key Laboratory of Animal Ecology and Conservation Biology,Institute of Zoology,Chinese Academy of Sciences,Beijing 100101,China
3 University of Chinese Academy of Sciences,Beijing 100049,China
4 College of Ecology,Lanzhou University,Lanzhou 730000,Gansu,China
Abstract eDNA metabarcoding is an advanced method for monitoring biodiversity proposed in recent years.By analyzing DNA in water,soil and sediment samples,the technology obtains species distribution and population quantity information.It was found that macrobarcode technology is more accurate than the traditional method in measuring the species richness of some groups.In Europe,America and South America,the reliability of this technology in monitoring amphibian diversity in the wild was studied,and it was found to be better than traditional biodiversity monitoring methods in detecting species diversity.At present,amphibian monitoring mainly depends on various traditional methods,such as transects,drift fence traps,artificial shelters and mark-recapture.These monitoring techniques have many shortcomings,such as low accuracy and strong subjectivity of study results.These technologies have poor effects on rare,invasive and endangered species with strong concealment ability,low density and strong seasonality and are difficult to implement in sites inaccessible to people.Traditional monitoring technology also requires considerable investment of human and material resources,and the economic cost is relatively high,while eDNA metabarcoding ismore efficient and less costly,so it is important to use eDNA metabarcoding in amphibian monitoring in China.In this study,the eDNA metabarcoding and traditional line transect method (TLTM) were used to study the characteristics of the two methods in the Beijing-Tianjin-Hebei region.Repeated sampling was conducted on 58 waterbodies in July 2019 and June 2020.After sequencing the samples using highthroughput sequencing technology,the differences between metabarcoding and commonly used TLTM surveys in detecting the diversity of four amphibians in North China were assessed.Our results showed that eDNA metabarcoding is more sensitive to the detection of the four amphibian species in the sampling area,and the combined use of eDNA metabarcoding and TLTM can improve the survey results of amphibians in the survey area to the greatest extent.In addition,in the process of species classification and identification of metabarcoding results,7 species of reptiles were detected,indicating that eDNA metabarcoding is also useful to detect reptiles.The results of this study indicate that metabarcoding in combination with TLTM can accurately estimate the diversity of amphibians in a short-term survey in North China and is also useful in reptile species detection.
Keywords anura,biodiversity,eDNA,metabarcoding
1.Introduction
In the past few hundred years,with the increase in human activities and the rate of species extinction,the loss of global biodiversity has become one of the most critical environmental problems,and amphibians are one of the most vulnerable taxa in the world (Hoffmannet al.,2010).In this case,biodiversity monitoring is particularly important for biodiversity protection.Biodiversity surveys essentially monitor changes in biodiversity over a period of time.The information obtained can provide a scientific basis for the development trend of biodiversity assessment and future biodiversity supervision strategies.The traditional line transect method (TLTM) is currently being used as a conventional technique for biodiversity assessment and long-term monitoring in terrestrial and aquatic ecosystems.Compared with infrared cameras and drift fences,this method has a lower environmental impact and time investment(Burnhamet al.,1980).However,traditional biodiversity monitoring methods based on acoustic or visual sampling are often limited (Yateset al.,2019;Deineret al.,2017;Thomsen and Willerslev,2015).Researchers need a method that can detect uncommon,unobservable,invasive and endangered species.This method should have the characteristics of high accuracy,low cost and no environmental impact.
Environmental DNA (eDNA) is DNA that has been released by an organism into the environment via feces,fur,urine,skin,gametes,etc.This DNA can be extracted from environmental samples,such as soil,water,and feces,without having to isolate the target organism.It is composed of intracellular DNA and extracellular DNA.The eDNA method is based on the fact that all species leave DNA traces in their environment,and this DNA can be collected.After collection,the DNA is extracted from the samples and analyzed using polymerase chain reaction (PCR) or quantitative PCR (qPCR) with a speciesspecific oligo system.After amplification in the PCR process,the amplified fragments are subsequently sequenced using a next-generation sequencing platform.The resulting sequences are then compared with a reference database to establish a list of species whose DNA is present in the sample.This approach is called eDNA metabarcoding.This method has been widely used in microbiology for many years,but it has just emerged and gained a firm foothold in the evaluation of amphibians and fish (Deineret al.,2015;Thomsen and Willerslev,2015;Valentiniet al.,2016;Liet al.,2019;Liet al.,2021a;Wanget al.,2022).The application of metabarcoding in the ecosystem is not only useful to describe the community and biodiversity but also to detect the interaction between populations on a large spatial scale.However,it may be limited by false reads caused by contamination or other errors (Ficetolaet al.,2016;Fonseca,2018;Bohmannet al.,2014).In other words,compared with traditional sampling methods,metabarcoding improves the detection speed,accuracy and identification ability and reduces the cost.However,at present,it needs to be standardized and unified and to combine taxonomy and molecular methods for complete ecological research (Gibsonet al.,2015;Liet al.,2021b).
Amphibians are one of the most vulnerable taxa in the world (Hoffmannet al.,2010).The main ecological conservation areas and important water sources in northwestern Beijing,Tianjin and Hebei were selected in this study as the research sites.These research sites play an important ecological role in water and soil conservation in the Beijing-Tianjin-Hebei region.At present,there is an urgent need for effective methods to evaluate and monitor amphibian diversity beyond the TLTM(Burnhamet al.,1980) because there are many deficiencies in its practical application,such as the need for researchers to have sufficient ability to identify species and the fact that surveys conducted in concealed places can only rely on call recognition and counting.Using metabarcoding combined with TLTM to monitor amphibian diversity will help to complement traditional methods.Traditional monitoring technology also requires considerable investment of human and material resources and the economic cost is relatively high,while eDNA metabarcoding is more efficient and less costly,so it is important to use eDNA metabarcoding in amphibian monitoring in China.Therefore,this study selected 58 waterbodies in the Beijing-Tianjin-Hebei region for multiple TLTM and eDNA surveys (Table S1).The effectiveness of metabarcoding in the monitoring of amphibian diversity in North China was tested and compared with that of TLTM.The distribution of amphibians in the Beijing-Tianjin-Hebei region was also evaluated through two investigation methods,and some suggestions for protection are provided.
2.Materials and Methods
2.1.Survey area and methodologyThe Beijing-Tianjin-Hebei region is an important monitoring area of China’s ecological environment.This study selected the main ecological conservation areas and important water sources in northwestern Beijing,Tianjin,and Hebei as the research sites.These research sites play an important ecological role in maintaining water and soil resources in the Beijing-Tianjin-Hebei region.The relationship between soil types and vegetation distribution in the area is clear,showing distinct horizontal and vertical zonal characteristics with the change in bioclimatic zone and mountain altitude.The vegetation type in the area is mainly deciduous broad-leaved forest.
This study used eDNA metabarcoding and TLTM to investigate the diversity of amphibians in the study area in mid-July 2019 and June 2020.In each grid,survey plots were set up according to the main habitat types of amphibians.The sample plots covered different habitat types,altitudes and ecological functional areas,taking into account historical records of amphibian distribution in the survey area.Although the eDNA primer was developed for amphibians,it can also detect reptiles,so we also investigated reptiles in the study area(Figure S1,Table S2).
2.2.TLTM samplingSampling lines were set up for amphibians according to their suitable habitat types.The amphibian sampling line was mainly set at the water–land intersection (1 m on the left and right sides of the water line) from 19:30 to 23:30 every night.Two investigators simultaneously record the species,number of individuals and environmental factors (such as vegetation coverage and human disturbance).During the observation,the investigators moved at an average speed of 1 km/h without disturbing the amphibians in the sampling line and counted all the amphibian individuals and tadpoles that had not completed metamorphosis observed within the sampling zone.During sampling,investigators recorded the number of amphibian species detected by visual observation and supplemented it with the number of amphibians detected from calls in the transect lines to avoid counting the same individuals in the same line during the simultaneous use of acoustic and visual observation.In this study,only the individuals of the same species who are seen or whose calls are heard in the same zone are identified as effective individuals.In addition,different calls are identified as different individuals.If no biological individuals are observed,they are also recorded as effective individuals.We also used the TLTM to survey reptiles in the study area.
2.3.Reference DNA databaseIn this study,eight amphibian samples collected during TLTM investigation and specimens stored in the laboratory were used to construct the local amphibian reference database.Due to the limited sample types,some reptile 12S RNA sequences in the NCBI database were also used (Table S3).We constructed a reference database of the 12S RNA mitochondrial sequences as completely as possible to ensure appropriate taxonomic assignment to sequences recovered from the environmental samples.We sequenced at least one individual from each of the species that was found using the traditional survey method and preserved it in 96% ethanol.As it was a challenge to collect samples from all expected species,we supplemented our local amphibian sequence reference database with sequences available from the EMBL database (release 143).Following the extraction of toe clip tissues (1–3 individuals per species) from amphibian species,we amplified the 12S rRNA mitochondrial gene in a dedicated room.We sequenced the PCR products using Sanger sequencing(Baiet al.,2012;Wanget al.,2014).
We downloaded all vertebrate sequences from the EMBL database.From our local reference database and the EMBL database,we extracted the relevant fragment of 12S sequences for metabarcoding analyses using the programs ecoPCR 0.5.0(Ficetolaet al.,2010) and OBITools 1.1.22 (Boyeret al.,2016).The final 12S metabarcoding reference database is as complete as possible given the available tissues and EMBL accessions.For the 28 species that were detected in our aquatic eDNA samples,we used the final 12S metabarcoding reference database to attribute taxonomy to the sequences.
2.4.eDNA samplingIn July 2019 and June 2020,58 sampling points were sampled.During sampling,the investigators collected water samples between 19:30 and 23:30 every night for metabarcoding analysis.Before the TLTM investigation,five locations were randomly selected along the waterbody.At each location,110 mL of surface water was collected with sterile bottles previously sterilized in the laboratory.After mixing five 110 mL samples of surface water,a 550 mL water sample (one water sample) was finally obtained.
On the night of sampling,we used a 1.5-μmol/L glass fiber(GF) filter membrane to filter the water samples.To control for possible contamination of the water sample caused by contamination from the equipment or experimental operational problems during transportation and for possible cross-contamination among waterbodies when extracting different water samples from different waterbodies,1000 mL ultrapure water was filtered through a filter membrane at each waterbody every night to establish a negative control.Subsequently,the filter membrane was stored separately in a 1.5-mL screw-top microcentrifuge tube filled with ethanol.During the filtration process,the laboratory personnel closed the doors and windows.When using tweezers to clamp the filter membrane,care was taken not to touch its center.The filter membrane was meticulously aligned to prevent sample contamination during the filtration process.All tubes were brought back to the laboratory and stored at −20 °C until DNA was extracted.To prevent cross contamination of water samples,10% commercial bleach (approximately 0.5%hypochlorous acid) was used to remove the DNA attached to the bottle and filtration equipment before each filtration of water samples and between filtrations of different water samples.Then,the bottle was cleaned with DNA-free distilled water.The personnel collecting and filtering the water samples wore new latex gloves at the time of sampling every day and night.
PCR and sequencingWe extracted mitochondrial DNA(mtDNA) from the filters of each water sample using a General AllGen Kit (Beijing CoWin Biotech Co.,Ltd.,Beijing,China) (Heet al.,2011),which generated 100 µL of DNA extract.We amplified DNA from a short fragment of the 12S rRNA mitochondrial gene in a mixture that included 2 µL of DNA extract as a template,1 U of Start Taq DNA Polymerase,0.2 mmol/L of each dNTP,0.2 IM of the batra_F (5’-ACACCGCCCGTCACCCT-3’)and batra_R (5’-GTAYACTTATGTTACTT-3’) primers and 4 µM of the human blocking primer batra_blk(5’-TCACCCTCCTCAAGTATACTTCAAAGGCA-SPC3I-3’)(Valentiniet al.,2016).The “batra” primers were used for the amplifications because they have a high proportion of sequences related to amphibian and fish species (99.28%) and show high taxonomic discrimination (Valentinet al.,2016).The primers (batra_F and batra_R) were 5’ labeled with a unique six-nucleotide tag (with at least three differences between tags),so we could differentiate all the samples and assign sequences to the individual samples during the sequence filtering process based on the primer tags.The following PCR program was employed: denaturation at 94 °C for 5 min;followed by 35 cycles of 30 s at 94 °C,30 s at 58 °C and 30 s at 72 °C;and a final elongation step at 72 °C for 10 min.The PCR was run for 9 replicates per sample.Additional negative controls were generated using ddH2O (instead of the filters) during DNA extraction and PCR.According to a previous study (Dejeanet al.,2012),we used DNA extracted fromLithobates catesbeianusas a positive control to verify whether the amplification of DNA from the target species in the eDNA samples failed due to inhibition of the PCR (Goldberg,2016).We ran parallel PCR assays of the 348 negative (including 174 negative field controls and 174 PCR processing controls) and 174 positive controls.Amplification and DNA extraction were performed in isolated rooms.
Before preparing the libraries,we examined the quality of each PCR product based on fluorescent signals using gel electrophoresis.All the negative controls (negative field controls and PCR processing controls) showed a lack of PCR products following gel electrophoresis.Each PCR product was pooled in equal volume to construct a library for the expected sequencing depth of 300 000 reads per sample.We prepared libraries using the DNA Sample Prep Reagent Set (MyGenostics,Beijing,China),which included end-repair and adapter ligation.We built 13 libraries for the PCR products from the water samples,with each library having 32 samples (a total of 416 samples,which included 408 water samples from this study and 8 from other projects),two libraries for the negative controls (one for the PCR control and one for the field control) and one library for the positive controls.The DNA for libraries was further purified before sequencing using SPRI beads according to the manufacturer’s protocol.The enrichment libraries were sequenced on an Illumina HiSeq X Ten sequencer with pairedend reads of 150 bp.
Bioinformatics analysisWe filtered and annotated the eDNA sequence reads using functions implemented in the OBITools package (Boyeret al.,2016).We assembled direct and reverse strands to construct consensus sequences and then removed low-quality sequences from the consensus sequences(FASTQ average quality score <35) using the Illumina pairedend program.We identified each sequence record by its corresponding sample based on its molecular tag (no errors allowed) and assigned primers (2-bp errors per primer allowed)to PCR products using the “ngsfilter” function.Unaligned sequences were removed from the datasets with the “obigrep”function.For each library,we clustered strictly identical sequences into operational taxonomic units (OTUs) using the“obiuniq” function to keep the information from the read counts.Using the “obigrep” function,we removed sequences with lengths that were too long (>130 bp) or too short (<30 bp),deleted OTUs with read counts ≤10,and retained only the OTUs with total read counts >10 for the denoising analysis.We labeled the sequences in each library as “head”,“singleton”,or“internal” (most likely due to amplification/sequencing errors and chimeric sequences) using the “obiclean” function (Boyeret al.,2016).We then removed the “singleton” and “internal”sequences from each library and retained only the “head”sequences for taxonomic assignment (options–r 0.05 -H) (Balintet al.,2018).We downloaded the gene sequence data from the EMBL database (release 143),NCBI;these data included information on different taxonomic groups (vertebrates,mammals,prokaryotes,fungi,invertebrates and plants) and environmental samples.We constructed the NCBI database of 12S rRNA mitochondrial sequences based on the downloaded data using ecoPCR (Ficetolaet al.,2010).Sequences from the samples and negative and positive controls were assigned taxonomy according to the NCBI reference database and local amphibian reference database using the ecotag tool.Sequences were assigned to a database sequence based on a 95% similarity threshold.
2.5.Statistical analysisWe compared the detection probability of species obtained by eDNA sampling with that obtained by traditional sampling.For each sample,we calculated the detection probability of species detected by each method.Specifically,we divided the number of sampling sites detected by one method by the total number of sampling sites.
In the whole sampling area,we used the dilution curve to fit the relationship between the expected species richness of the eDNA method and TLTM and the expected sampling workload.We compared the dilution curves between TLTM and metabarcoding for each sample.We also fitted the sampling workload required by the two methods to observe all species in all study waterbodies.In the “vegan” package of R language version 3.3.2,the “rich” and “rarc” functions were used for 2000-random-simulation dilution analyses.All statistical analyses were performed using R 3.3.2 (R 2015).The significance level was set atP<0.05.
3.Results
Using the TLTM method,a total of 4 species of amphibians were found,includingBufo gargarizans,Rana chensinensis,Kaloula borealis,andPelophylax nigromaculatus,which belonged to 1 order,3 families and 4 genera and accounted for 50% of the species recorded in the history of the survey area.R.chensinensiswas distributed in 63.8% of the sampling sites.B.gargarizanswas distributed in 63.8% of the sampling sites.P.nigromaculatuswas distributed in 46.6% of the sampling sites.K.borealiswas distributed in 10% of the sampling sites.
Using the TLTM method,a total of 8 species of reptiles were found that belonged to 1 order,5 families and 7 genera and accounted for 48% of the species recorded in the history of the survey area.These reptiles includedGekko swinhonis,Eremias brenchleyi,Dinodon rufozonatum,Elaphe davidi,Elaphe dione,Elaphe taeniura,Elaphe mandarina,andGloydius intermedius.Eremias brenchleyiis distributed in 51.7% of the sampling sites.Dinodon rufozonatumis distributed in 10% of the sampling sites.Elaphe davidi,Elaphe dione,Elaphe taeniura,Elaphe mandarinaandGloydius intermediuswere found in only one survey area.
A total of 408 water samples were collected from 58 sampling points,excluding the negative control and positive control.After the screening process,12 219 400 reads were obtained from 408 samples.The range was 170–346 986 reads per sample,and the average sequence number of each sample was 29 949 reads (Table S4).
Four amphibian species were identified after comparison with the local amphibian reference database (Bufo gargarizans,Rana chensinensis,Kaloula borealis,andPelophylax nigromaculatus).According to the classification results of the local reptile reference database,a total of 7 species of reptiles were identified.
The metabarcoding results showed that the species occupancy rates ofRana chensinensisandBufo gargarizanswere the highest at 70.6% and 75.8%,respectively.Pelophylax nigromaculatusexhibited an occupancy rate of 51.7% and that ofKaloula borealiswas 15.5% (Figure 1).Comparing the results of TLTM and metabarcoding at each sampling point,it was found that the number of species investigated by the two methods was the same for 63% of the sampling points.In the remaining 37% of the samples,metabarcoding detected more species than the TLTM method.
Thettest showed that the detection probability of metabarcoding for each species (P≤ 0.007) and all combined species (P<0.0001) was greater than that of the TLTM (Table 1).In all waterbodies,the average species richness detected by metabarcoding was greater than that detected by TLTM(Kruskal–Wallis test,χ2=56.05,P<0.0001) .
The sparse curve shows that the sampling efforts required by the two methods to detect species richness in the whole sampling area are very different (Figure 2).The waterbodies only needed to be sampled 20–30 times for metabarcoding analysis to obtain a good estimate of all four amphibian species.In contrast,there were only four species close to the horizontal asymptote in the 58 waterbodies when sampled by the TLTM.To obtain a better estimation using the TLTM method,more samples should be collected.Using TLTM to sample 58 waterbodies during only three visits yields values that are close to but still slightly lower than the estimates for all four amphibian species (Figures 2–3).
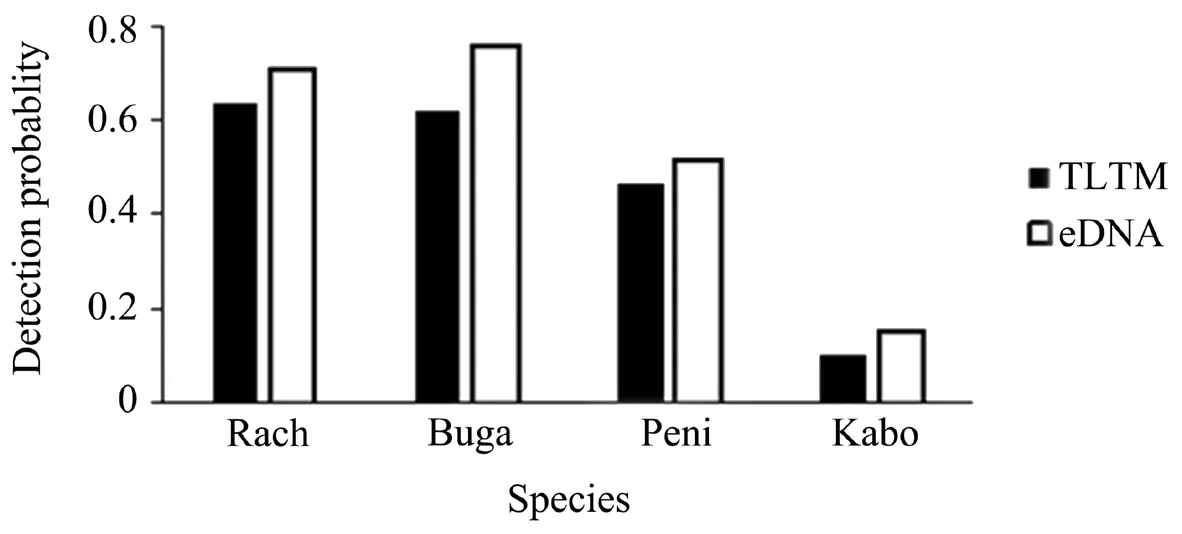
Figure 1 Overall detection probability of anuran species according to the two methods.Rach=R.chensinensis;Buga=B.gargarizans;Peni=P.nigromaculatus;Kabo=K.borealis.
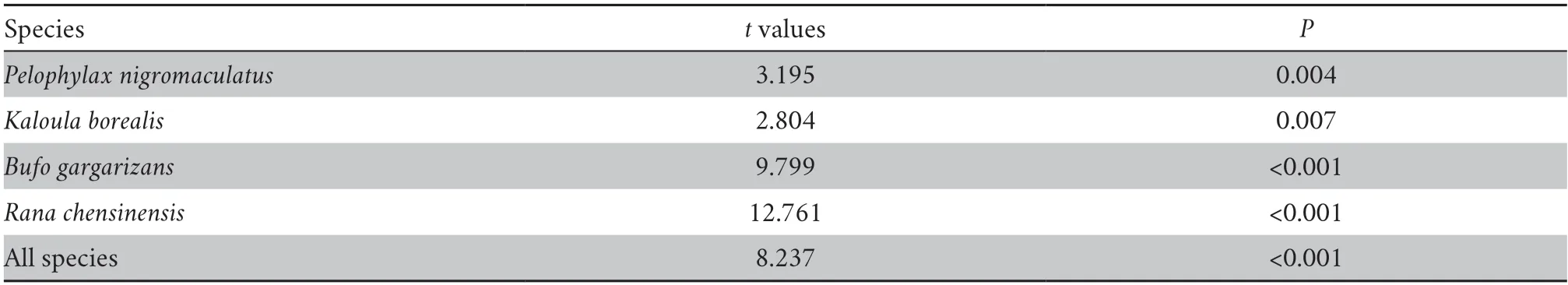
Table 1 Student’s t test of differences in the detection probability of anuran species detection probability between eDNA metabarcoding and TLTM.
4.Discussion
The TLTM results show that in terms of species number,only 50% of amphibian species found in historical records have been found in the Beijing-Tianjin-Hebei region.In terms of distribution,Rana chensinensisis the amphibian with the widest distribution range in the survey area,followed byBufo gargarizans.After comparing the species detected in this study with those in historical records,we found that not all species in the historical record were detected,which may be due to the small number of sampling sites surveyed in Hebei and to the fact that the sampling sites did not include the habitats of some species,such asH.japonica.Another explanation is that in this study,only 1–5 transect lines were established at the sampling points,and only two investigators participated in the surveys.Due to insufficient investigation,no rare and hidden amphibian species,such asStrauchbufo raddeiandL.catesbeiana,were detected in the area.As an invasive species,L.catesbeianais mostly released during religious practices or escapes during the trade process.In this study,bullfrog release sites or flower,bird,fish,insect or vegetable markets were not included as sampling sites,soL.catesbeianawas not detected in this study.

Figure 2 Rarefaction curves of species richness based on the number of waterbodies sampled using the two methods.
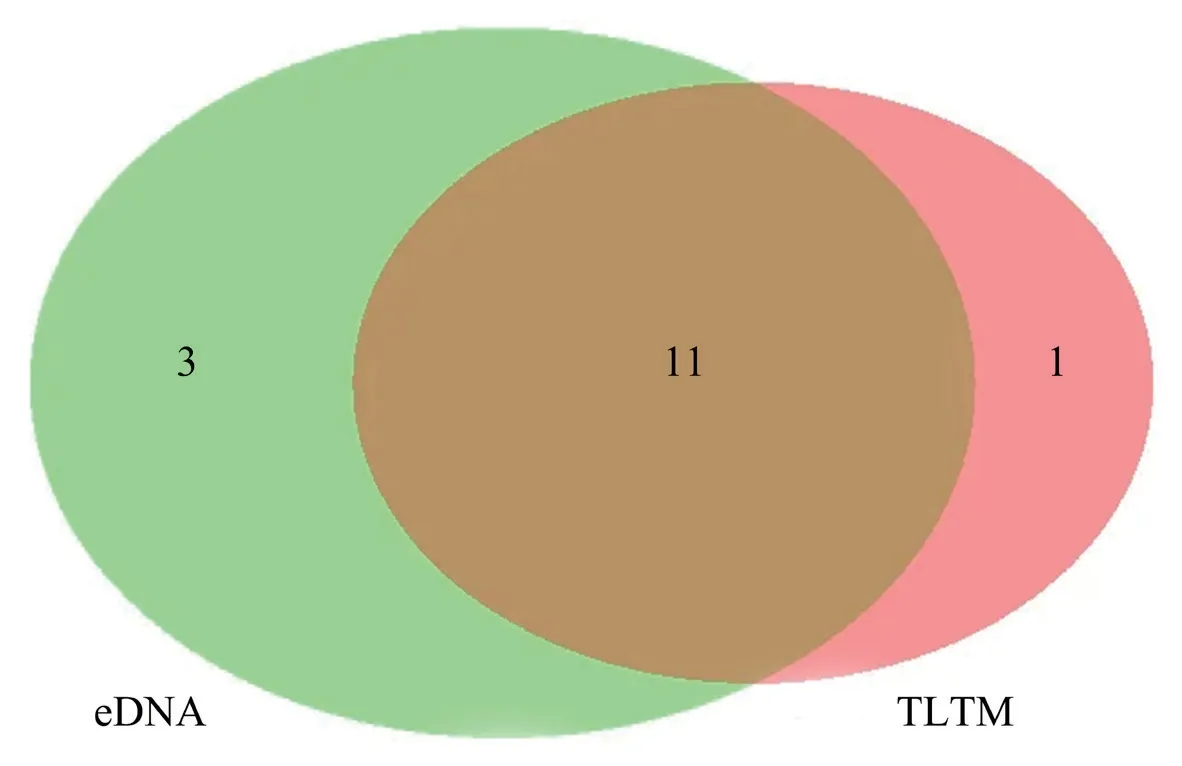
Figure 3 Number of species detected by eDNA and TLTM.
Through large-scale repeated sampling,this study compared and tested the effects of metabarcoding and TLTM in the detection of amphibian diversity in North China.In the detection of amphibian diversity,metabarcoding exhibits an increased detection probability for all species compared to TLTM,and eDNA has shown a better result than many traditional methods in biodiversity detection (Statet al.,2019;Singeret al.,2019;Cristescuet al.,2018;Deineret al.,2016).Metabarcoding detected the presence of all four amphibian species in the study area,which is the same as that detected according by TLTM.eDNA can be a supplementary means in biodiversity detection (Cilleroset al.,2019;Pontet al.,2018;Raphaet al.,2016;Portet al.,2016).However,in the statistical results for single sampling points,metabarcoding detected more species than TLTM in 63% of the sampling points.In terms of the number of species,we found only 50% of the amphibian species found in historical records in the Beijing-Tianjin-Hebei region.In terms of distribution,the relative read count and occurrence rate of species in the metabarcoding results were the same as those detected in the TLTM survey.Rana chensinensisis the most widely distributed amphibian in the survey area,followed byBufo gargarizans.The distribution range of these species is the same as that recorded in historical data records.
In addition,for each waterbody,metabarcoding detected the occurrence of each amphibian species identified by the TLTM.Historical data showed thatStrauchbufo raddeistill existed in Beijing,but this species was not detected by either method.One explanation is that there is no habitat forBufo gargarizansin the sampling area,and the sampling area we designated does not include the distribution area ofStrauchbufo raddei.Another explanation is that both methods sample only very limited areas or habitats in the waterbody.Due to limited sampling effort or the low density ofStrauchbufo raddei,this species may not be found in the whole sampling area.
Metabarcoding previously detected seven types of reptiles,which showed the potential of metabarcoding in detecting biodiversity in addition to fish and amphibians (Calvignac-Spenceret al.,2013;Boessenkoolet al.,2012;Aylagaset al.,2015;Bohmannet al.,2014).For each waterbody,metabarcoding also basically detected the occurrence of each species identified by TLTM.Eremias arguswas only detected by eDNA.Gloydius intermediusandElaphe taeniurawere detected only by TLTM.It may be that reptiles,especiallyGloydius intermediusandElaphe taeniura,prefer to inhabit hillsides,plains,hills and other terrain and are common in rivers,grass and rice fields.During eDNA sampling,most of the sampling points selected in this study were in calm waters,such as ponds and reservoirs,and there was no sampling in flowing waterbodies,such as streams and rivers.
Metabarcoding was found to have a higher detection probability for each amphibian species than TLTM,which may be because TLTM is unable to accurately sample these amphibians.The TLTM is one of the best traditional methods for the rapid assessment of biodiversity (Rosenzweig,1995).Density and abundance can differ greatly from TLTM estimates depending on habitat type,habitat complexity,ecology and behavior of a species,prevailing weather,season,and the ability and experience of researchers (Ribeiro-Júnioret al.,2008).Metabarcoding results cannot reflect the age and sex ratio of species.Therefore,in species investigations,eDNA can be used as a supplement to traditional methods,such as the TLTM.After using eDNA for species detection at different sampling points,the TLTM method can be used to conduct supplementary sampling and increase the number of samples in the future to ensure the credibility of the final survey results.
AcknowledgementsThis study is supported by grants from The Biodiversity Survey and Assessment Project of the Ministry of Ecology and Environment,China(2019HJ2096001006),National Natural Science Foundation of China (32030070),the High-Level Talents Research Start-Up Project of Hebei University,China’s Biodiversity Observation Network (Sino-BON),and Second Tibetan Plateau Scientific Expedition and Research (STEP) Program (2019QZKK0501).
Appendix
The Figure S1 and Tables S1–S4 can be downloaded from the website https://pan.baidu.com/s/1ynpI11qVkUhKaXVr8T7tJg (access code:1234).
杂志排行

Asian Herpetological Research的其它文章
- Desertification Drives the Shift in Egg Size-Number Trade-Off in an Agamid Lizard
- Endocast Morphological Variation and Its Driving Forces in Scutiger boulengeri
- Metagenomic Analysis of Mangshan Pit Viper (Protobothrops mangshanensis) Gut Microbiota Reveals Differences among Wild and Captive Individuals Linked to Hibernating Behaviors
- Lineage Diversification and Niche Evolution in the Chinese Cobra Naja atra(Elapidae)
- Phylogenetic Relationships among Chinese Rice Frogs within the Fejervarya limnocharis Species Complex (Amphibia: Dicroglossidae)
- Three New Species of Diploderma Hallowell,1861 (Reptilia: Squamata:Agamidae) from the Shaluli Mountains in Western Sichuan,China