美登素衍生物DM1聚合物杂质聚合物1的合成研究
2022-12-16朱阳姚长亮夏广兵
朱阳 姚长亮 夏广兵
(上海医药集团股份有限公司中央研究院 上海 201203)
DM1 是 N2´-去乙酰基 -N2´-(3-巯基 -1-氧代丙基)美登素的英文简称。DM1是一种能够抑制细胞分裂的微管蛋白抑制剂,其与曲妥珠单抗通过偶联方式形成靶向抗肿瘤药物T-DM1[1],后者的商品名为Kadcyla,2013年2月获得美国FDA批准,用于治疗人表皮生长因子受体-2阳性的晚期乳腺癌患者。
在DM1的制备过程[2-4]中,会产生一种DM1聚合物“聚合物1”(图1)。聚合物1是DM1制备过程中产生的杂质,故须在DM1成品中予以严格控制。制备聚合物1并进行相关研究,对于选择合适方法有效控制甚至去除此杂质,进而提高DM1乃至T-DM1的质量具有重要的意义。
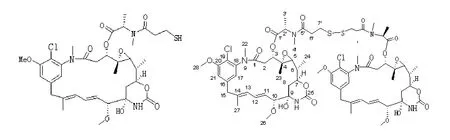
图1 DM1(左)和聚合物1(右)的化学结构
有文献[2]报告了聚合物1的制备方法:将DM1溶于由磷酸缓冲液和乙醇组成的混合液中,在2, 2'-二硫二吡啶存在的条件下使DM1发生脱氢聚合生成聚合物1,最后通过浓缩、萃取、洗涤、减压蒸馏等,残余物再用色谱柱分离制得纯聚合物1。我们按文献操作,发现收率非常低,制备困难。对此,我们通过大量实验摸索,发现DM1在1, 3-二甲基-2-咪唑啉酮(1, 3-dimethyl-2-imidazolidinone, DMI)或N, N-二甲基甲酰胺(N,N-dimethylformamide, DMF)等有机溶剂中很容易发生二聚化而生成聚合物1。利用该法制备聚合物1,操作简便且易获得高纯度的聚合物1,经检索未见有相似研究报告。
1 仪器与试剂
1.1 仪器
Bruker Ascend 400 MHz核磁共振波谱仪(nuclear magnetic resonance spectroscopy, NMR)(内标:四甲基硅烷);Agilent 1260高效液相色谱仪(high performance liquid chromatography, HPLC)等。
1.2 试剂
DM1(自制,合成路线见图2,HPLC检测纯度>99%);安丝菌素P-3(ansamitocin P-3, AP3)(HPLC检测纯度>95%)、DMI(HPLC检测纯度>98%)等试剂均为市售分析纯试剂。
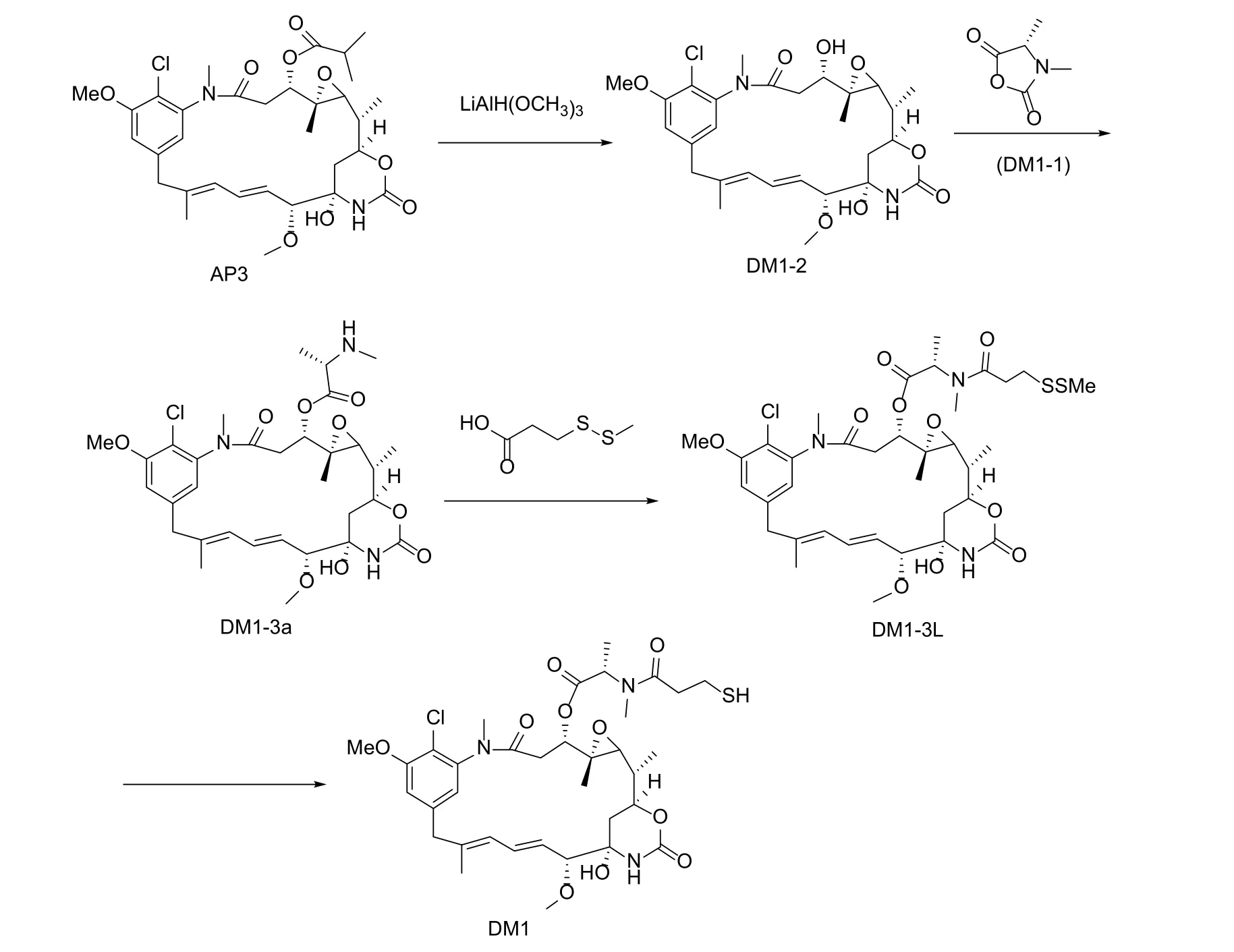
图2 DM1合成路线
2 制备方法
2.1 DM1的制备
2.1.1 DM1-2制备
在反应瓶中加入500 mL四氢呋喃,氩气保护下压入300 mL 1 mol/L四氢铝锂的四氢呋喃溶液,冷却至<-45 ℃后慢慢滴加30 g无水甲醇,内温控制在-45 ~-35 ℃,制得LiAl(OMe)3待用。将10.0 g(15.76 mmol)AP3溶于200 mL四氢呋喃中,然后快速滴加至上述LiAl(OMe)3中,氩气保护,于-45 ~ -35 ℃反应1.5 h。反应结束后,加入150 mL饱和氯化铵水溶液淬灭反应,再加入300 mL乙酸乙酯进行萃取,分出有机相,加入无水硫酸镁脱水干燥1 h左右。过滤,(30±5)℃下减压蒸馏,得8.8 g(15.57 mmol)泡状固体即DM1-2,收率98.8%。
1H-NMR(400 MHz, CDCl3):δ 7.18(d, J=1.5 Hz,1H),6.88(d, J=1.5 Hz, 1H),6.51(s, 1H),6.49(dd,J=11、15 Hz, 1H),6.13(d, J=11 Hz, 1H),5.50(dd,J=9、15 Hz, 1H),4.40(d, 1H),4.09(m, 1H),3.96(s,3H),3.46(d, 1H),3.44 ~ 3.45(d, 2H),3.33(s, 3H),3.18(s, 3H),3.12(d, J=12 Hz, 1H),2.58(d, J=9 Hz,1H),2.28(dd, 1H),2.09(dd, 1H),1.65(s, 3H),1.54(m,1H),1.25(d, 3H),1.22(d, 2H),0.79(s, 3H)。
1H-NMR、13C-NMR和质谱检测均确证所得物质为DM1-2。
2.1.2 DM1-3a制备
在反应瓶中分别加入9.6 g(74.42 mmol)DM1-1、8.5 g(15.01 mmol)DM1-2和 130 mL DMF, 氩 气保护下搅拌溶解。控制温度<10 ℃,分批加入催化剂Zn(CF3SO3)2(共18 g)和N, N-二异丙基乙胺(共15 g),15 ~ 20 ℃搅拌反应过夜。翌日,加入200 mL饱和碳酸氢钠水溶液淬灭反应,然后加入200 mL饱和氯化钠水溶液,搅拌5 ~ 10 min,再用500 mL乙酸乙酯萃取共3 ~ 5次。分出并合并有机相,用500 mL水和500 mL饱和氯化钠水溶液分别洗涤后,于(30±5)℃、0.08 MPa条件下减压蒸馏至干,得12.5 g DM1-3a粗品。以柱层析纯化,采用二氯甲烷-甲醇(20∶1)洗脱。收集产物流分,(30±5)℃下减压蒸馏至干,得8.7 g(13.38 mmol)黄色固体即DM1-3a,收率89.1%。
1H-NMR(400 MHz, CDCl3):δ 6.87(d, J=1.5 Hz,1H),6.85(d, J=1.5 Hz, 1H),6.43(dd, J=11、15 Hz,1H),6.14(d, J=11 Hz, 1H),5.52(dd, J=9、15 Hz,1H),4.94(dd, J=3、12 Hz, 1H),4.26(m, 1H),3.98(s,3H),3.69(q, 1H),3.50(d, 1H),3.49(d, 1H),3.37(s, 3H),3.23(dd, 1H),3.12(s, 3H),2.59(d, J=12 Hz, 1H),2.54(d, J=9 Hz, 1H),2.37(s, 3H),2.19(dd,J=3、14 Hz, 1H),1.67(s, 3H),1.58(d, J=13 Hz, 1H),1.38(m, 1H),1.30(d, 3H),1.27(d, 3H),1.24(d,1H),0.84(s, 3H)。
1H-NMR、13C-NMR和质谱检测均确证所得物质为DM1-3a。
2.1.3 DM1-3L制备
在反应瓶中分别加入7.5 g(11.54 mmol)DM1-3a和60 mL二氯甲烷,搅拌溶解后再依次加入7.6 g 3-(甲基二硫烷基)丙酸和8.8 g 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,室温搅拌反应3.0 h。然后,依次向反应瓶中加入60 mL pH为6的磷酸缓冲液和30 mL二氯甲烷,搅拌,分出有机相,水相用30 mL二氯甲烷萃取。合并有机相,用水洗涤2次后于(30±5)℃下减压蒸馏至干,得10.5 g DM1-3L粗品。粗品用50 mL乙醇重结晶2次,得5.1 g(6.50 mmol)白色固体即DM1-3L,收率56.3%。
1H-NMR(400 MHz, CDCl3):δ 6.82(d, J=1.5 Hz,1H),6.73(d, J=11 Hz, 1H),6.64(d, J=1.5 Hz, 1H),6.44(dd, J=11、15 Hz, 1H),6.30(s, 1H),5.66(dd,J=9、15 Hz, 1H),5.42(q, J=7 Hz, 1H),4.77(dd, J=3、12 Hz, 1H),4.26(m, 1H),3.98(s, 3H),3.69(d, J=12 Hz, 1H),3.46(s, 1H),3.55(d, J=9 Hz, 1H),3.35(s,3H),3.23(s, 3H),3.12(d, J=12 Hz, 1H),3.09(d,J=9 Hz, 1H),2.90(m, 2H),2.88(s, 3H),2.85(m,2H),2.60(m, 1H),2.26(s, 3H),2.17(dd, J=3、14 Hz, 1H),1.98(s, 1H),1.64(s, 3H),1.55(d, J=13 Hz, 1H),1.46(m, 1H),1.23 ~ 1.31(m, 6H),1.21(d,J=13 Hz, 1H),0.80(s, 3H)。
1H-NMR、13C-NMR和质谱检测均确证所得物质为DM1-3L。
2.1.4 DM1制备
将5 g(6.37 mmol)DM1-3L粗品和5.1 g DL-二硫苏糖醇用由120 mL乙酸乙酯和120 mL甲醇组成的混合液溶解后,加入120 mL pH为7.5的磷酸缓冲液,氩气保护下室温反应3.5 h。然后,加入240 mL pH为6.0的磷酸缓冲液,用120 mL乙酸乙酯萃取,有机相用30 mL水洗涤5次,再于(30±5)℃下减压蒸馏至干,得5.2 g DM1粗品。以柱层析纯化,采用二氯甲烷-甲醇(20∶1)洗脱。收集产物流分,(30±5)℃下减压蒸馏至干,得4.1 g(5.55 mmol)白色固体即DM1,收率87.1%,HPLC检测纯度>99%。
1H-NMR(400 MHz, CDCl3):δ 6.86(d, J=1.5 Hz,1H),6.75(d, J=11 Hz, 1H),6.69(d, J=1.5 Hz, 1H),6.46(dd, J=11、15 Hz, 1H),6.29(s, 1H),5.66(dd,J=9、15 Hz, 1H),5.45(q, J=7 Hz, 1H),4.78(dd, J=3、12 Hz, 1H),4.26(m, 1H),3.99(s, 3H),3.69(d, J=12 Hz, 1H),3.55(d, J=9 Hz, 1H),3.46(s, 1H),3.37(s,3H),3.22(s, 3H),3.12(d, J=12 Hz, 3H),3.04(d,J=9 Hz, 1H),2.86(s, 3H),2.60 ~ 2.83(m, 5H),2.19(dd,J=3、14 Hz, 1H),1.78(s, 1H),1.68(s, 3H),1.58(d,J=13 Hz, 1H),1.43 ~ 1.63(m, 1H),1.27 ~ 1.35(m,6H),1.24(d, J=13 Hz, 1H),0.81(s, 3H)。
1H-NMR、13C-NMR和质谱检测均确证所得物质为DM1。
2.2 聚合物1的制备
2.2.1 方法一
将1.5 g(2.03 mmol)DM1加至由40 mL DMI和40 mL水组成的的混合液中,室温搅拌20 h左右,至反应液中已逐渐有白色固体析出且HPLC检测显示DM1完全消失止。加入200 mL水,搅拌并冷却到0 ~ 5 ℃,1 h后过滤。滤饼用少量80%乙醇水溶液洗涤,然后真空干燥,得1.2 g(0.81 mmol)白色固体即聚合物1,收率79.8%,HPLC检测纯度>98%。
1H-NMR(400 MHz, 二甲基亚砜 -d6):δ 7.17(d,J=1.2 Hz, 1H),6.87(s, 1H),6.69 ~ 6.44(m, 3H),5.91(s, 1H),5.68 ~ 5.47(m, 1H),5.31(q, J=6.6 Hz, 1H),4.54(dd, J=2.5、12.0 Hz, 1H),4.08(t, J=11.8 Hz, 1H),3.94(s, 3H),3.50(d, J=9.0 Hz, 1H),3.45(d, J=12.4 Hz, 1H),3.27(s, 3H),3.18(d, J=12.5 Hz, 1H),3.10(s,3H),2.84 ~ 2.74(m, 2H),2.70(s, 3H),2.57 ~ 2.45(m,4H),2.05(d, J=12.1 Hz, 1H),1.60(s, 3H),1.46(d,J=12.9 Hz, 2H),1.27(d, J=3.4 Hz, 1H),1.18(d, J=6.8 Hz, 3H),1.13(d, J=6.3 Hz, 3H),0.79(s, 3H)。
电喷雾电离质谱:[M+H]+计算值为1 473.53,实测值是1 473.56。
1H-NMR、13C-NMR、DEPT、COSY、HSQC、HMBC和质谱检测均确证所得物质为聚合物1。
2.2.2 方法二
将100 mg(0.136 mmol)DM1加至4 mL DMF中,搅拌加热至80 ℃反应31 h。然后,冷却到室温,加入20 mL水,继续搅拌冷却到0 ~ 5 ℃。1 h左右后过滤,滤饼用水洗涤,得100 mg白色固体即聚合物1粗品。以1 cm×10 cm硅胶柱纯化,采用二氯甲烷-甲醇(20∶1)洗脱。收集产物流分,于(30±5)℃、0.08 MPa条件下减压蒸馏至干,得65 mg(0.044 mmol)白色固体即聚合物1,收率64.7%,HPLC检测纯度为95%。
3 结果与讨论
本研究是在研究DM1制备过程中对DM1的聚合物杂质聚合物1进行的合成研究。我们通过实验摸索发现,DM1在DMI、DMF等有机溶剂中很容易发生二聚化而生成聚合物1,尤其是在选用市售DMI为溶剂时,经过简单操作,不需繁琐的后处理或柱层析纯化,即能获得高纯度的聚合物1,可直接用作DM1有关物质质量控制的对照品。此外,这一发现也提示我们,欲在DM1制备过程中有效控制甚至避免聚合物1的产生,必须在DM1制备过程中尽可能不使用DMI、DMF等有机溶剂,此有利于获得高纯度的DM1及其产业化。
致谢:感谢上海医药集团股份有限公司中央研究院的质量研发团队与所有参与此项目研究的同事们的大力支持。
