大豆分离蛋白- 单宁酸- 多糖三元复合物的功能特性及结构表征
2022-12-15皇甫云鹏包怡红赵鑫磊骆嘉原姜喆卉
皇甫云鹏, 包怡红,2,*, 赵鑫磊, 骆嘉原, 姜喆卉
(1.东北林业大学 林学院, 黑龙江 哈尔滨 150040;2.黑龙江省森林食品资源利用重点实验室, 黑龙江 哈尔滨 150040)
大豆为我国主要的农作物之一。大豆分离蛋白(soybean protein isolate,SPI)是大豆的主要工业化产品,蛋白质含量高于90%[1],因其富含多种必需氨基酸且氨基酸组成合理,被公认为优质植物蛋白[2]。SPI在食品、医药和化工领域中发挥着重要作用[3],但因其溶解性、乳化性和起泡性等功能性质较差,在一定程度上限制了其应用。因此,通过改变SPI的结构进而改善其功能特性,成为近年来的研究热点。蛋白质改性常用的方法有化学改性(糖基化、磷酸化、乙酰化、亲脂化、巯基化和交联等)、物理改性(高压处理、高压均质和高强度超声等)和酶改性(谷氨酰胺转氨酶、氧化酶和内肽酶等)[4-6],相比之下,化学改性更有效且成本更低[7-9]。
研究发现,通过碱处理、自由基接枝法制备的蛋白质- 多酚共价复合物以及通过糖基化制备的蛋白质- 多糖共价复合物的功能特性都优于改性前的蛋白质[10-11]。接枝多酚引入的羟基对蛋白质- 多酚共价复合物的抗氧化能力有较大的贡献,Guo等[12]的研究发现单宁酸与SPI在碱性条件下可以形成共价键,增加了SPI的抗氧化活性。多酚的共价接枝同样可以改变蛋白质的界面性质,Sui等[13]的研究报道,SPI与黑米花青素之间的共价和非共价结合均改善了蛋白质的乳化和起泡特性,并且SPI-花青素共价复合物比非共价复合物具有更好的起泡性。蛋白质- 多糖共价复合物含有较多的亲水基团和更为疏松的表面结构,从而具有更好的溶解性和乳化活性[14]。多糖的共价接枝也可以增强蛋白质的远距离空间排斥,提高其乳化稳定性和泡沫稳定性[15-16]。Hu等[17]发现SPI-杏鲍菇多糖共价复合物的乳化活性和乳化稳定性在pH值3~11内均显著优于天然SPI,共价复合物作为乳化剂稳定β-胡萝卜素乳液时,多糖产生的空间位阻可以促进乳液中功能因子的包埋,并减少油滴聚集。因此利用多酚和多糖对蛋白质进行共价修饰,可以开发具有较好功能性质的新型食品级材料。蛋白质- 多酚- 多糖三元共价复合物可将蛋白质的乳化活性、多酚的抗氧化活性和多糖的空间位阻效应有机地结合起来[18],具有更加优异的功能特性。肌原纤维蛋白(myofibrillar protein,MP)-葡聚糖(dextran,DEX)共价复合物与不同多酚(表没食子儿茶素没食子酸酯、儿茶素和没食子酸)的共价接枝均降低了其二级结构中α-螺旋的含量,3种三元共价复合物的热稳定性和抗氧化活性均显著优于MP-DEX[7]。Liu等[19]研究绿原酸- 乳铁蛋白- 葡聚糖三元共价复合物,发现三元共价复合物表现出更好的热稳定性和乳化性能,可作为抗氧化性乳化剂更有效地提高β-胡萝卜素乳液的物理稳定性并抑制β-胡萝卜素的化学降解。
目前,多酚、多糖、SPI组成的三元共价复合物的研究还存在很多空白。为了使改性后的SPI能兼具多酚和多糖两种物质的优点、获得更加理想的功能特性,本研究采用富含羟基的水溶性多酚——单宁酸(tannin acid,TA),以及水溶性较好的多糖——麦芽糊精(maltodextrin,MD)和聚葡萄糖(polydextrose,PD)对SPI进行改性,在碱处理法制备SPI-TA的基础上,采用湿法美拉德反应将MD、PD分别共价接枝到SPI-TA上,制备三元共价复合物SPI-TA-MD、SPI-TA-PD,并且与2种SPI-TA的三元物理混合物进行对比,对其结构和性质进行分析,探究多酚和多糖的共价接枝对SPI结构和功能性质的影响。本研究旨在为SPI的改性提供新的思路和理论依据,以期提高其在食品工业中的应用价值。
1 材料与方法
1.1 材料与试剂
SPI(w≥98%),购自上海源叶生物科技有限公司;TA(w≥99%),购自上海阿拉丁生化科技股份有限公司;MD(w≥99%,DE=18)、PD(w≥90%,分子量162~15 000,其中5 000以内的占88.7%),购自河南万邦化工科技有限公司;氢氧化钠,购自北京化工厂;透析袋(截留分子质量3 kDa)、福林酚、L-亮氨酸、丙烯酰胺,购自北京博奥拓达科技有限公司;十二烷基磺酸钠(sodium dodecyl sulfate,SDS),购自北京索莱宝科技有限公司;邻苯二甲醛(OPA),购自天津市大茂化学试剂厂;蛋白Marker(5~250 kDa),购自赛默飞世尔中国;松籽油,购自吉林省辉庆农业开发有限公司,试剂均为国产分析纯。
1.2 仪器与设备
PHS-3C型实验室pH计,上海仪电科学仪器股份有限公司;PL203型电子分析天平,日本岛津公司;79-1型磁力加热搅拌器,山东博科再生医学有限公司;FD-1A-50型冷冻干燥机,北京博医康实验仪器有限公司;TENSOR27型傅里叶红外光谱仪,德国Bruker公司;DSC Q20型差示扫描量热仪,美国TA仪器;LS55型荧光分光光度计,珀金埃尔默股份有限公司。
1.3 实验方法
1.3.1SPI-TA共价复合物的制备
参考Guo等[12]的方法制备SPI-TA共价复合物。将SPI和TA按照质量比10∶1溶解在去离子水中,使溶液中SPI的质量分数为2%。将溶液放置在4 ℃环境中过夜以充分水合,使用浓度为0.5 mol/L的NaOH溶液将其pH值调节至10.0,并暴露于空气中搅拌反应24 h。利用3 kDa的透析袋将反应后的溶液在4 ℃条件下透析48 h,每隔8 h换一次水以确保除去未参与反应的游离TA。将透析液在-20 ℃下预冻6 h,进行真空冷冻干燥48 h,得到SPI-TA共价复合物粉末。
1.3.2三元共价复合物的制备
参考Weng等[20]的方法通过湿法美拉德反应制备SPI-TA-MD、SPI-TA-PD共价复合物。将已充分水合,质量分数为6%的MD、PD水溶液分别与质量分数为2%的SPI-TA溶液等体积混合,混合溶液中SPI-TA与MD、PD的质量比为1∶3,在恒温95 ℃条件下加热并搅拌反应1 h后,迅速冷却至室温结束反应,用浓度为1 mol/L的HCl溶液和NaOH溶液调节体系的pH值至7.0,预冻后真空冷冻干燥得到三元共价复合物,记为SPI-TA-MD con、SPI-TA-PD con。相同条件下,不加热制备SPI-TA与MD、PD的三元物理混合物溶液,预冻后真空冷冻干燥得到三元物理混合物,记为SPI-TA-MD mix、SPI-TA-PD mix。
1.3.3反应基团的测定
1.3.3.1 多酚接枝量的测定
参考Xu等[7]的方法测量样品中的多酚含量。将1.0 mL质量浓度为10 mg/mL的样品溶液与2.5 mL福林酚试剂混合,并在25 ℃避光反应 5 min,加入2 mL质量分数为7.5%的Na2CO3溶液并将混合物避光2 h以进行反应。使用酶标仪在760 nm波长处测量最终溶液的吸光度,采用TA标准品绘制吸光值- 多酚浓度的标准曲线,TA的接枝量按式(1)计算。
(1)
式(1)中,b(TA)为TA接枝量,μmol/g;ρ1为TA的质量浓度,mg/mL;ρ2为样品中SPI的质量浓度,mg/mL;M为TA的摩尔质量,1 701.20 g/mol。
1.3.3.2 游离巯基含量的测定
参考李杨等[21]的方法,将15 mg样品溶于5 mL Tris-甘氨酸缓冲液(86 mmol/L的Tris,90 mmol/L的甘氨酸,4 mmol/L的EDTA,8 mol/L的尿素,质量分数为1%的SDS,pH值8.0)中配置样品溶液。将 4 mg 的5,5-二硫硝基苯甲酸(DNTB)溶解于1 mL的Tris-甘氨酸缓冲液中,取上述溶液50 μL与样品溶液涡旋混合均匀,室温反应1 h,以未加蛋白质的混合液为空白,在412 nm波长处测量其吸光度A412。巯基含量的计算见式(2)。
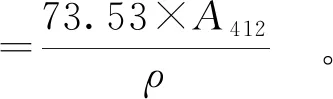
(2)
式(2)中,b(巯基)为巯基质量摩尔浓度,μmol/g;1.36×104为摩尔消光系数;73.53=106/(1.36×104);ρ为样品中SPI的质量浓度,mg/mL。
1.3.3.3 游离氨基含量及多糖接枝度的测定
参考赵城彬等[22]方法,将80 mg的OPA溶解在2 mL的甲醇中,分别加入质量分数为20%的SDS溶液5.0 mL、浓度为0.1 mol/L的硼砂溶液50 mL及200 μL的β-巯基乙醇,最后用去离子水定容到100 mL配置成OPA试剂。将样品粉末溶解在去离子水中,配制成质量浓度为5 mg/mL的溶液。测定时,将4 mL OPA试剂和200 μL样品溶液充分混合,然后在35 ℃水浴中反应2 min,使用酶标仪在340 nm波长处测定吸光值A340,根据吸光值计算样品中游离氨基含量,以赖氨酸为标准品作标准曲线。三元共价复合物中MD、PD的接枝度(DG)的计算见式(3)。
(3)
式(3)中,C0为SPI-TA分别和MD、PD物理混合物的自由氨基含量,μmol/mg;Ct为三元共价复合物自由氨基的含量,μmol/mg。
1.3.4蛋白质电泳分析
参考Laemmli[23]的方法进行SDS-聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)实验,对蛋白及其共价复合物进行凝胶电泳分析。将冻干样品分散到去离子水中,配制成质量浓度为2 mg/mL的样品溶液,分离胶、浓缩胶中丙烯酰胺质量分数为12%、5%,每孔上样量为10 μL,采用5~250 kDa的蛋白Marker,电泳之后通过考马斯亮蓝R250对凝胶条带染色后进行分析。
1.3.5紫外-可见光吸收光谱分析
参考Liu等[10]的方法,准确称量不同冻干粉,用去离子水配制成蛋白质质量浓度为1.0 mg/mL的溶液。通过酶标仪在200~450 nm波长下扫描样品,速度为1 nm/s,采用去离子水作为空白参照。
1.3.6傅里叶红外光谱的测定
采用溴化钾压片法测定样品的红外光谱。将1 mg样品与100 mg溴化钾混合研磨成均匀粉末,在傅里叶红外光谱仪上扫描4 000~500 cm-1波数内的红外光谱(FTIR),采用溴化钾作为空白对照。
1.3.7溶解性的测定
参考Klompong等[24]的方法分析蛋白质及其共价复合物的溶解性。将200 mg的冻干样品分散在20 mL去离子水中,配成质量浓度为10 mg/mL的溶液,采用浓度为0.1 mol/mL的HCl、NaOH溶液将体系的pH值调节至7.0,室温下搅拌30 min,8 000 r/min离心20 min。采用考马斯亮蓝法测定上清液中的蛋白质质量浓度。同时将样品在浓度为0.5 mol/L的NaOH溶液中溶解,测定样品中的总蛋白质质量浓度。蛋白质的溶解性根据式(4)计算。
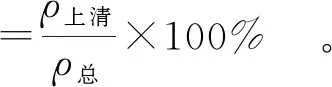
(4)
式(4)中,ρ上清为上清液中的蛋白质质量浓度,mg/mL;ρ总为总蛋白质质量浓度,mg/mL。
1.3.8抗氧化活性的测定
1.3.8.1 ABTS+自由基清除率测定
参考Zhou等[25]的方法,测定样品的ABTS+自由基的清除率。取浓度为7 mmol/L的ABTS+水溶液与浓度为2.45 mmol/L的过硫酸钾溶液混合,避光反应12 h以上制备得到ABTS+溶液,实验前用甲醇将ABTS+溶液稀释,使其在747 nm波长处的吸光度值为0.7±0.020。取质量浓度为0.2 mg/mL的样品溶液1 mL,加入3 mL ABTS+溶液,振荡混匀,避光反应60 min,在734 nm波长处测定吸光值。运用式(5)来计算ABTS+自由基清除率。
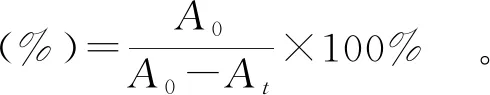
(5)
式(5)中,A0为空白对照的吸光值;At为反应60 min 后反应液的吸光值。
1.3.8.2 DPPH自由基清除率测定
参考Gong等[26]的方法测定样品的DPPH自由基清除率。用乙醇配制浓度为1.75×10-4mol/L的DPPH溶液,取质量浓度为0.2 mg/mL的样品溶液2 mL加入到2 mL的DPPH乙醇溶液中,在室温下避光反应1 h,随后检测其在517 nm波长处的吸光值,根据式(5)来计算DPPH自由基清除率。
1.3.8.3 还原能力测定
参考Yi等[27]的方法测定样品的还原能力。取2 mL的样品溶液(质量浓度为0.2 mg/mL,溶解在体积分数为2%的醋酸溶液中),加入1 mL的铁氰化钾溶液(质量浓度为10 mg/mL),振荡混匀后,于50 ℃水浴反应20 min,随后加入1 mL的三氯乙酸溶液(质量浓度为100 mg/mL),振荡混匀。取1 mL的上述溶液,加入3 mL的去离子水和0.4 mL的三氯化铁溶液(质量浓度为1 mg/mL),于室温下反应5 min,使用酶标仪检测其在700 nm处的吸光值,以吸光值大小表示其还原能力大小。
1.3.9乳化活性和乳化稳定性的测定
参考肖志刚等[28]的方法测定样品的乳化活性和乳化稳定性。冷冻干燥后的样品用去离子水分散后配置成质量浓度为2 mg/mL的溶液。按照溶液- 油体积比4∶1加入松籽油。采用高速剪切乳化机在10 000 rpm下剪切2 min后,移液枪移取底部100 μL的乳浊液,加入到5 mL质量分数为0.1%的SDS溶液中,于595 nm波长处测定吸光度,剪切后的乳液静置30 min后,按照上述操作再次测其吸光值。分别根据式(6)、(7)计算其乳化活性(emulsifying activity index,EAI)和乳化稳定性(emulsion stability index,ESI)。
(6)
(7)
式(6)、(7)中,EAI为乳化活性,m2/g;ESI为乳化稳定性,min;2为固定系数;2.303为反应速率常数;A0为初始吸光值;DF为稀释倍数(200);ρ为蛋白质的质量浓度,g/mL;L为光程,cm;α为油相占比;A30为静置30 min后的吸光值。
1.3.10起泡性和泡沫稳定性的测定
参考代世成等[29]的方法测定样品的起泡性及泡沫稳定性,使用高速剪切乳化机将15 mL质量浓度为3 mg/mL的蛋白质及其复合物溶液在10 000 r/min下剪切2 min。使用量筒来确定剪切前、剪切后0 min和剪切后静置30 min不同样品溶液的体积,分别根据式(8)、(9)计算样品的起泡性(foaming capacity,FC)和泡沫稳定性(foaming stability,FS)。
(8)
(9)
式(8)、(9)中,FC为起泡性,%;FS为泡沫稳定性,%;Vb为均质前的溶液体积,mL;V0为均质后 0 min 的溶液体积,mL;V30为均质后静止30 min的溶液体积,mL。
1.3.11表面疏水性的测定
参考Xu等[7]的方法,将8-苯胺基-1-萘磺酸钠(8-anilino-1-naphthalene sulfonate,ANS)溶于浓度为10 mmol/L的磷酸盐缓冲液中(pH值7.0),配置成浓度为8 mmol/L的ANS探针溶液。用磷酸盐缓冲液配置不同蛋白质质量浓度的样品溶液(0.05~0.2 mg/mL)。将样品溶液与ANS探针溶液按体积比200∶1混合均匀,利用荧光分光光度计在390 nm激发波长下测定其发射波长,计为样品的疏水性。
1.3.12热稳定性测定
参考Wang等[30]的方法,称取5.0 mg左右的样品放置在铝制的样品盒中并密封,以密封空铝盘作为空白对照,升温程序:30~180 ℃,升温速率为 5 ℃/min,干燥氮气的流速为50 mL/min。利用TA universal analysis 2000分析软件对得到的热曲线进行分析,计算出样品的变性温度。
1.4 数据处理
实验结果重复3次,采用Origin 8.0软件绘图,数据采用IBM SPSS Statistics 26软件进行单因素方差分析(ANOVA),P<0.05认为有显著性差异。
2 结果与分析
2.1 共价接枝对SPI基团的影响
SPI、SPI-TA、三元共价复合物和混合物中的巯基含量和自由氨基含量,见表1。由表1可知,通过福林酚法测定所制备的SPI-TA二元共价复合物的多酚结合当量为(35.56±1.32) μmol/g。碱性有氧条件可以促使TA中的酚羟基去质子化,酚羟基易被氧化成为相应的醌,进而与蛋白的亲核基团如游离氨基、赖氨酸、酪氨酸和半胱氨酸等反应,在酚环上形成C-N和C-S共价键,最终形成稳定的共价复合物[31]。

表1 SPI及其共价复合物和物理混合物的巯基含量、自由氨基含量、TA结合当量和多糖接枝度
游离巯基是蛋白质中最具活性的官能团之一,和蛋白质变性及其功能特性密切相关。由表1可知,与SPI相比,SPI-TA共价复合物的游离巯基含量显著下降(P<0.05),这可能是由于部分巯基被氧化成二硫键或是二硫键相互转换产生的[21]。此外,TA在碱性处理条件下产生的醌类物质也可直接与巯基发生相互作用形成C-S共价键,从而显著地降低SPI中巯基的含量[32]。SPI-TA-MD/PD三元共价复合物的巯基含量相比于物理混合物更高(P<0.05),这是因为湿法美拉德反应的条件会导致SPI的构象变化,反应过程中巯基基团逐渐暴露于SPI表面,因此巯基含量增加,这与Xu等[33]的研究结果相似。
评估自由氨基含量可以分析蛋白质-多酚共价复合物中多酚的结合程度,这种方法也常用来测定美拉德反应的进程。由表1可知,相比于SPI,SPI-TA共价复合物中自由氨基量减少15.15%,实验结果与Xu等[7]的研究相似。由于SDS可以破坏蛋白质和多酚间的非共价键,结果中的自由氨基含量降低表明,SPI与TA可能发生了共价结合,生成了C-N共价键[18]。
SPI-TA与多糖经过湿法美拉德反应后,SPI-TA-MD、SPI-TA-PD共价复合物中自由氨基都显著降低(P<0.05),因为美拉德反应发生时,蛋白质自由氨基和还原糖羰基通过共价连接形成席夫碱。SPI-TA-MD共价复合物中自由氨基的浓度低于SPI-TA-PD共价复合物(P<0.05),计算可得SPI-TA-MD共价复合物中MD的接枝度为46.54%,SPI-TA-PD共价复合物中PD的接枝度为32.26%,说明SPI-TA和MD之间美拉德反应程度更高。这种现象的产生主要由两种多糖的结构差异所致。一方面,PD是通过葡萄糖与山梨醇在柠檬酸的催化下真空熔融缩合制备,是高度分支的非消化葡萄糖聚合物,平均具有10或12个葡萄糖分子[34-35];而MD的平均聚合度为18,高于PD,因此在美拉德反应中当蛋白质和多糖之间的质量比相同时,MD具有比PD更多数量的C-末端分子,更易与SPI发生反应,因此接枝度更高。另一方面,MD水溶液的黏度比PD的更小,有利于蛋白质和多糖的美拉德反应[36]。有研究报道,糖的分子流动性可能是蛋白质与糖接枝反应具有不同反应速率的原因[37]。
2.2 共价接枝对SPI分子质量的影响
SPI及其共价复合物的SDS-PAGE结果如图1。由图1可知,泳道2中条带的迁移率不同,在35 kDa附近、50 kDa附近以及70~100 kDa 都有条带分布,分别对应着SPI的A亚基、β亚基、α和α′亚基部分[12]。泳道3、泳道4和泳道5在250 kDa以上有条带分布,表明二元复合物和三元复合物的分子量高于SPI,这是因为TA在反应过程中氧化成其相应的醌,并被SPI侧链中的亲核基团攻击,导致与SPI发生交联,形成大分子蛋白质聚集体[12, 38-39]。由于实验时使用的上样缓冲液中含有SDS和β-巯基乙醇,可以破坏蛋白质与多酚或多糖之间的非共价键,因此实验结果表明SPI和TA之间形成了共价键。并且从图1中可以看出SPI在经过TA修饰后,主要亚基部分的条带颜色也都明显变浅,这是因为相应蛋白质亚基参与了共价反应从而被消耗。通过对比泳道3、泳道4、泳道5中250 kDa分子质量以上的条带分布可以发现,泳道4和泳道5中高分子质量条带的颜色略浅于泳道3,这可能是因为MD、PD与SPI-TA共价结合消耗了部分SPI-TA,并且形成分子量更大的三元共价复合物聚集体[38]。
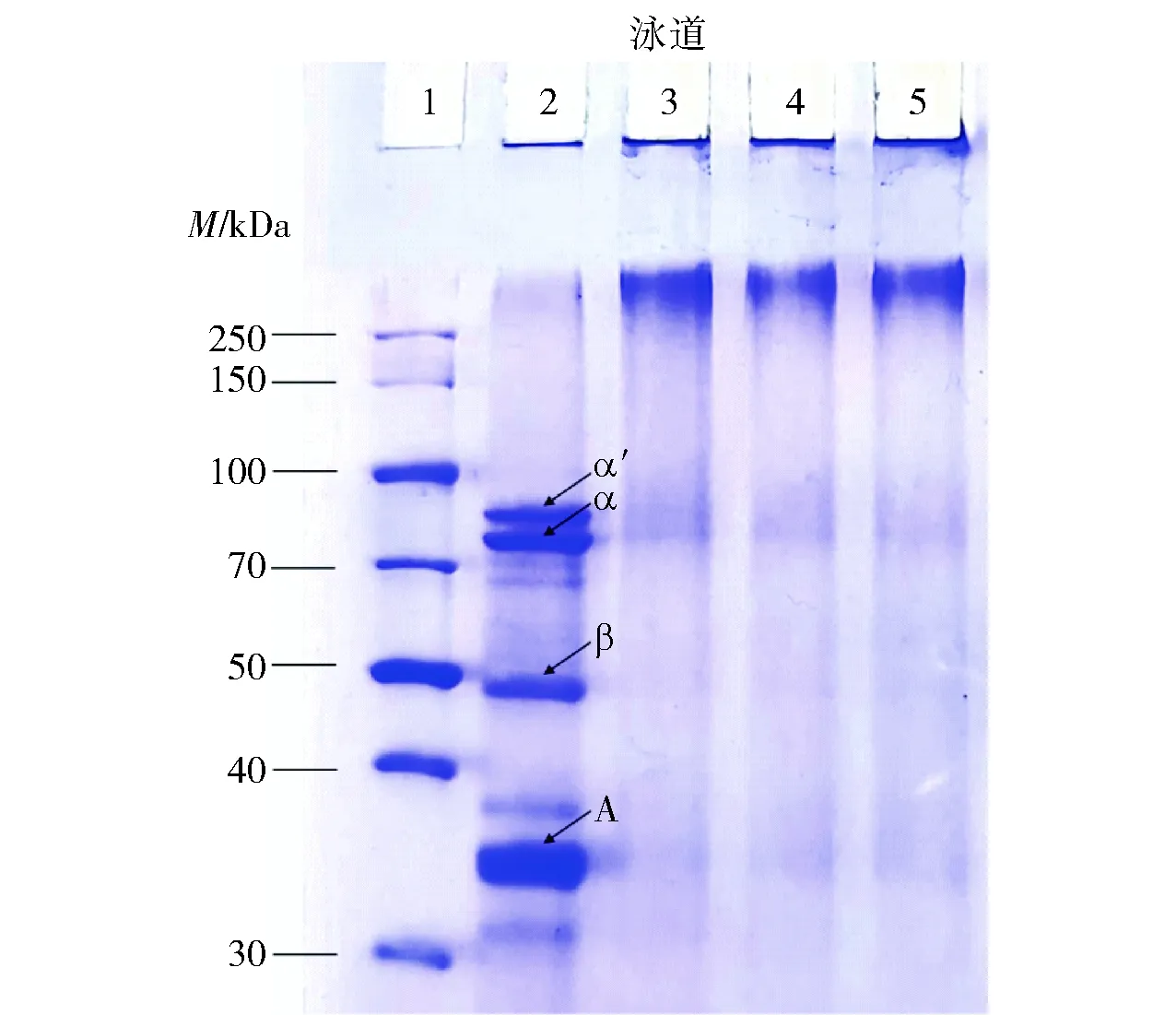
泳道1~5分别对应蛋白Marker、SPI、SPI-TA、SPI-TA-MD 共价复合物和SPI-TA-PD 共价复合物。α、α′、β和A为大豆分离蛋白的亚基。
2.3 共价接枝对SPI紫外- 可见光吸收光谱的影响
通过紫外- 可见光吸收光谱研究蛋白质和多酚的相互作用,SPI、SPI-TA、三元共价复合物和三元物理混合物的紫外- 可见光吸收光谱结果见图2。
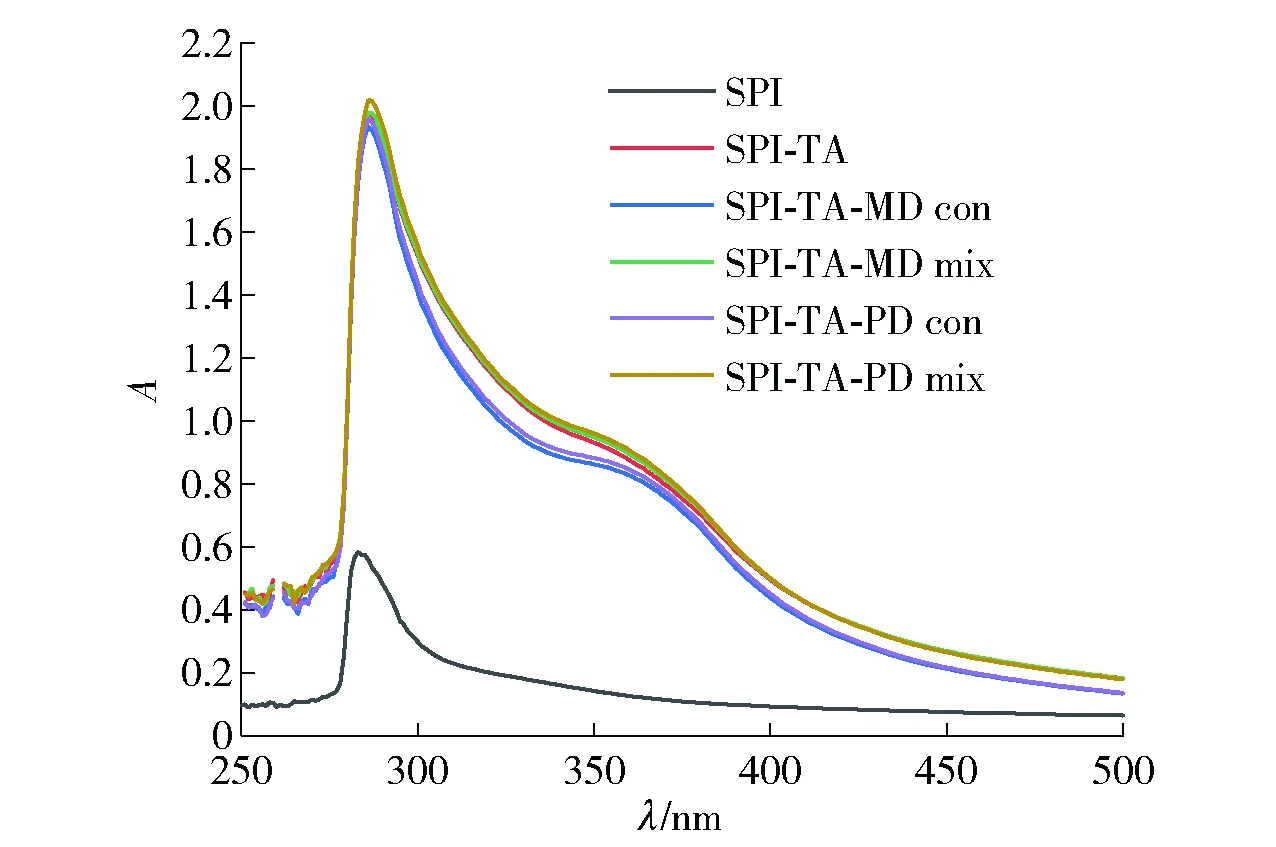
图2 大豆分离蛋白及其共价复合物和物理 混合物的紫外- 可见光吸收光谱
由图2可知,样品的吸光度均在285 nm波长处出现峰值,这是因为蛋白质中的一些氨基酸如酪氨酸、色氨酸和苯丙氨酸具有紫外吸收性质。SPI仅在283 nm处显示吸收峰,而SPI-TA和SPI-TA-MD、SPI-TA-PD共价复合物不仅在286 nm处具有最大吸收峰,而且在370 nm处出现了一个肩峰,肩峰的出现是因为SPI与TA的共价结合[10]。SPI共价接枝TA后,最大吸收峰波长红移,说明SPI的氨基酸残基的微环境发生了变化。并且SPI与TA、MD、PD的共价交联使蛋白质发生解折叠,导致色氨酸与酪氨酸残基暴露到了蛋白质的表面,使得吸光度增加。
2.4 共价接枝对SPI二级结构的影响

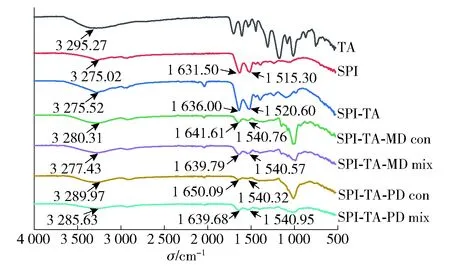
图3 TA、SPI、SPI-TA、SPI-TA-MD和SPI-TA-PD共价 复合物及物理混合物的FTIR光谱
据报道,多酚可以改变蛋白质的构象,主要是α-螺旋、β-折叠、β-转角和无规则卷曲之间的转换[42],蛋白质二级结构与酰胺Ⅰ带峰的对应关系是α-螺旋(1 646~1 664 cm-1)、β-折叠(1 615~1 637 cm-1和1 682~1 700 cm-1)、β-转角(1 664~1 681 cm-1)和无规则卷曲(1 637~1 645 cm-1)。利用peakfit软件对样品的酰胺Ⅰ带(1 600~1 700 cm-1)进行傅里叶变换、高斯去卷积可以大致得出蛋白二级结构的变化,如图4。由图4可知,接枝TA后,SPI的β-折叠结构减少了12.30%,α-螺旋、β-转角和无规则卷曲的比例分别增加了7.33%、12.09%、4.39%。这和吕思瑶等[43]关于SPI和染料木素共价作用的研究结果相似,说明TA对SPI造成了不可逆的结构改变。相比于SPI-TA以及相应的三元物理混合物,三元共价复合物表现出β-折叠结构进一步减少,α-螺旋和无规则卷曲含量进一步增加,这是因为MD、PD与SPI-TA 的共价结合导致空间结构的变化和蛋白质分子的展开。研究报道,蛋白质和多糖之间的美拉德反应会对蛋白质的二级结构造成影响,不仅是由于生物聚合物的相互作用,而且和湿法美拉德反应过程中蛋白质的热变性有关[44]。
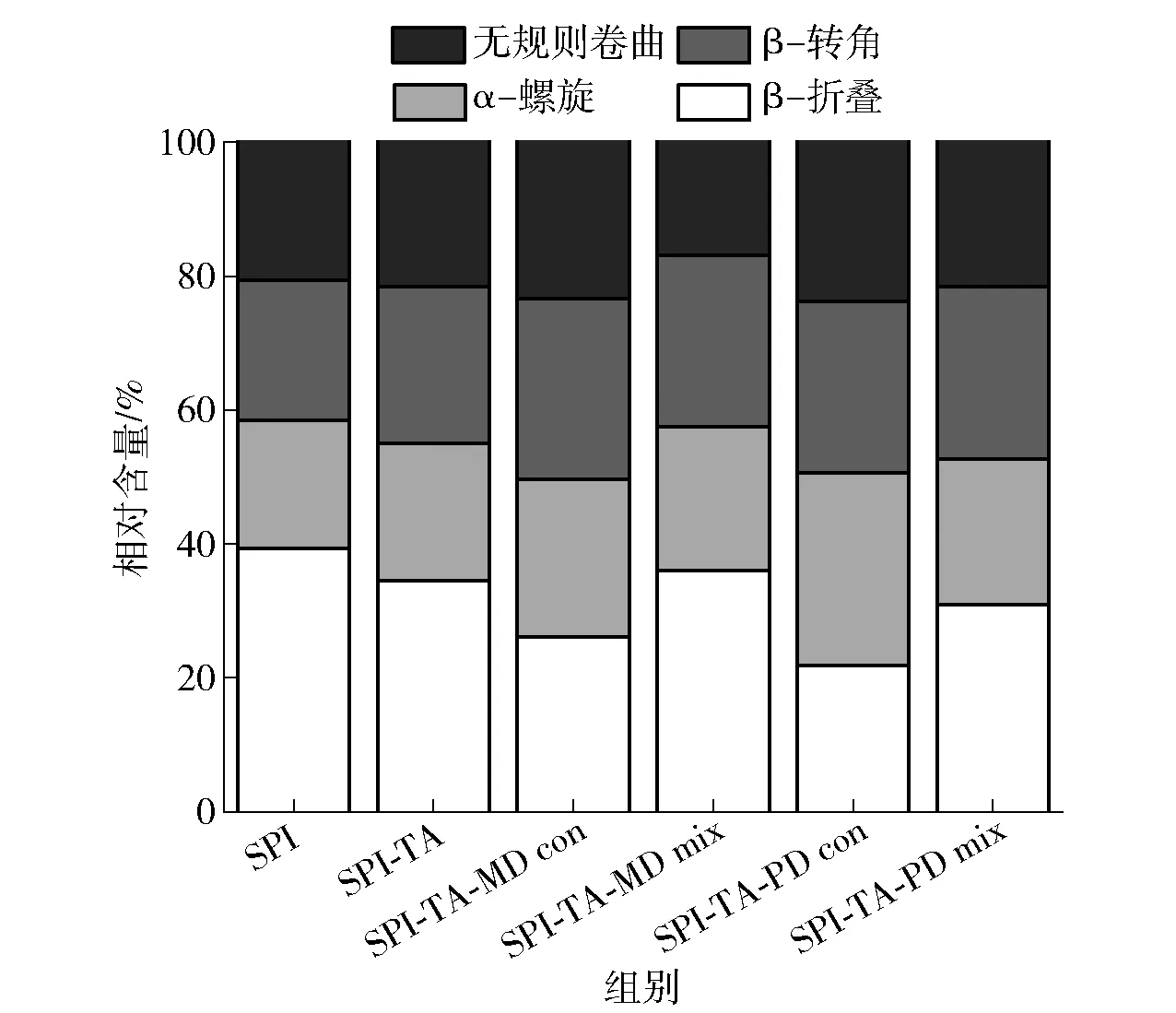
图4 SPI及其共价复合物和物理混合物的二级结构含量
2.5 共价接枝对SPI溶解性的影响
溶解性是蛋白质最重要的特性之一,会影响蛋白质的其他功能特性,例如乳化性[45]、胶凝特性[36]。植物蛋白溶解性差,限制了它们作为功能成分在药物和食品中的应用[46]。SPI及其共价复合物和物理混合物的溶解性如图5。由图5可知,SPI接枝TA后形成的SPI-TA共价复合物的溶解性显著下降。在共价反应的碱性条件下,TA氧化为醌类物质,与SPI侧链通过C-N键形成网状结构,从而形成了不溶性聚集体,其分子质量也有所增加。凝胶电泳结果(图1)也与这一结论相一致,这和代世成等[29]关于SPI和儿茶素共价复合物溶解性的研究结果相似。相比于SPI,SPI-TA-MD、SPI-TA-PD共价复合物的溶解性分别提高了32.17%、26.45%,这与反应的多糖有关。有研究发现在美拉德反应条件下,SPI的溶解性会提高[1, 47]。在美拉德反应初期,暴露在蛋白表面的自由氨基可以较快地与亲水性的MD、PD反应生成富含糖基的产物[48]。三元共价复合物比蛋白质本身或者三元物理混合物具有更多的亲水基团,产生的空间位阻可以阻止SPI分子之间的相互作用并提高SPI与水分子之间的亲和力[49-50],因此三元共价复合物具有更好的溶解性。
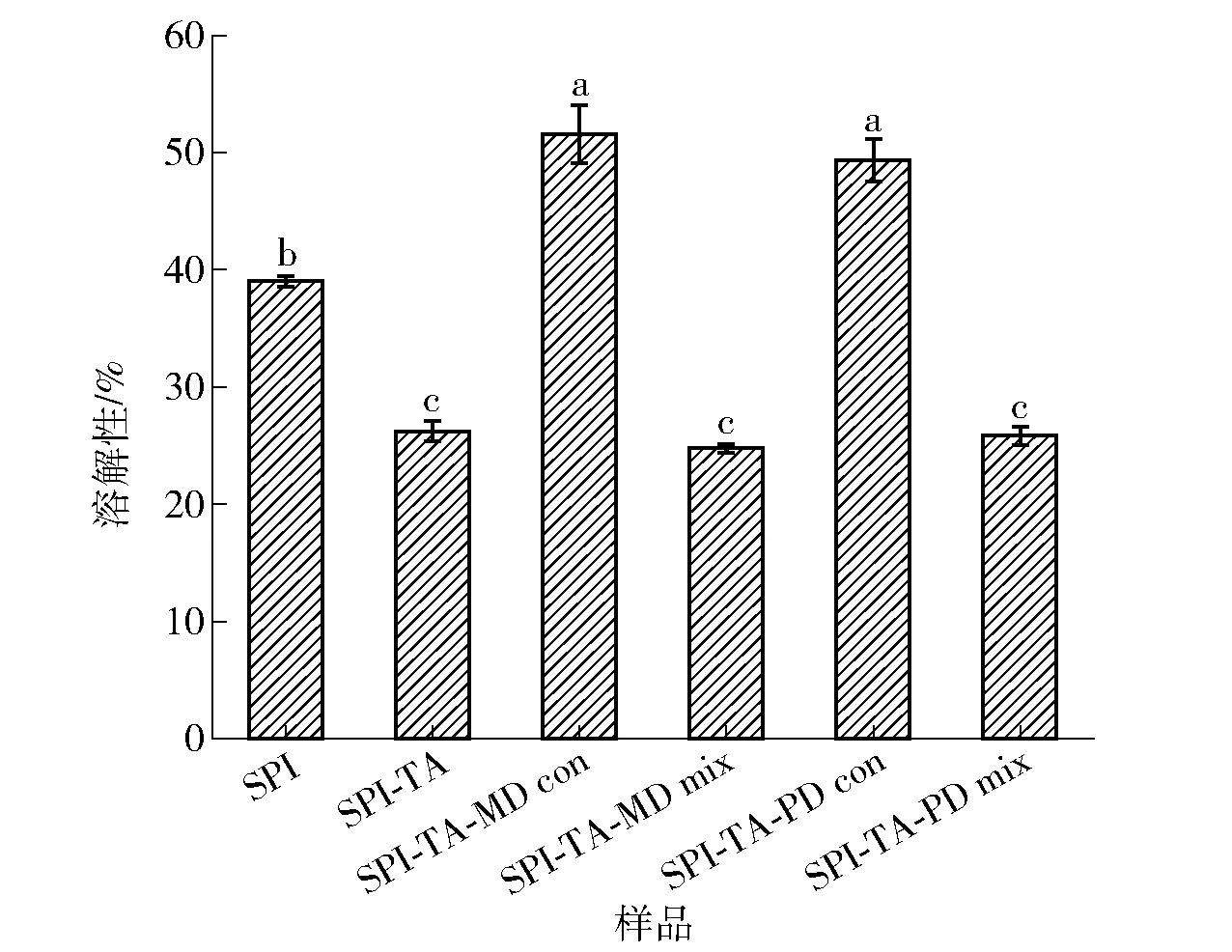
不同小写字母代表差异显著(P<0.05)。
2.6 共价接枝对SPI抗氧化活性的影响
因为不同抗氧化方法测定原理不同,单一的测定方法不能准确地评估活性物质的抗氧化能力。因此本研究分别以ABTS+自由基清除率、DPPH自由基清除率和Fe3+还原能力为指标,测定蛋白及其复合物的抗氧化活性,结果如图6所示。由图6可知,TA具有很强的抗氧化性,可作为抗氧化剂用于食品中,能够有效地延长食品的货架期。本研究中,SPI、TA、SPI-TA共价复合物、三元共价复合物和三元物理混合物的ABTS+自由基清除活性、DPPH自由基清除活性和Fe3+还原能力分别如图6所示。由图6可知,天然SPI的ABTS+自由基清除活性、DPPH自由基清除活性和还原能力较弱,而SPI-TA共价复合物显示出较高的抗氧化活性(P<0.05)。SPI-TA共价复合物的ABTS+自由基清除活性为66.36%,是SPI的3.41倍;SPI-TA共价复合物的DPPH自由基清除活性为71.53%,是SPI的8.50倍;SPI-TA共价复合物的还原能力是SPI的12.93倍。研究结果表明,SPI共价结合TA后形成的二元复合物具有强烈的自由基清除能力,能终止自由基链式反应。这主要是由于共价反应后SPI分子中引入了更多的羟基,羟基对物质的抗氧化能力有较大的贡献。同时二元和三元复合物的抗氧化能力低于相同浓度的TA溶液,这与蛋白和多酚的反应机制有关,两者的共价结合消耗了TA中部分的羟基。此外,空间位阻也在一定程度上导致共价结合的蛋白- 酚类化合物抗氧化能力的下降[51]。
由图6可知,2种三元共价复合物相比于SPI、SPI-TA以及它们的三元物理混合物具有更高的抗氧化活性。有研究表明,蛋白与还原糖通过美拉德反应可以得到具有抗氧化性的产物[52]。SPI-TA和多糖反应过程中生成的中间体还原酮化合物能够打破氢原子供给的自由基链,并被认为是自由基链反应的终止剂[53]。这可能是提高2种三元共价复合物的抗氧化活性的重要原因。由此可见,三元共价复合物可用作食品中有效的抗氧化剂,防止脂质氧化。
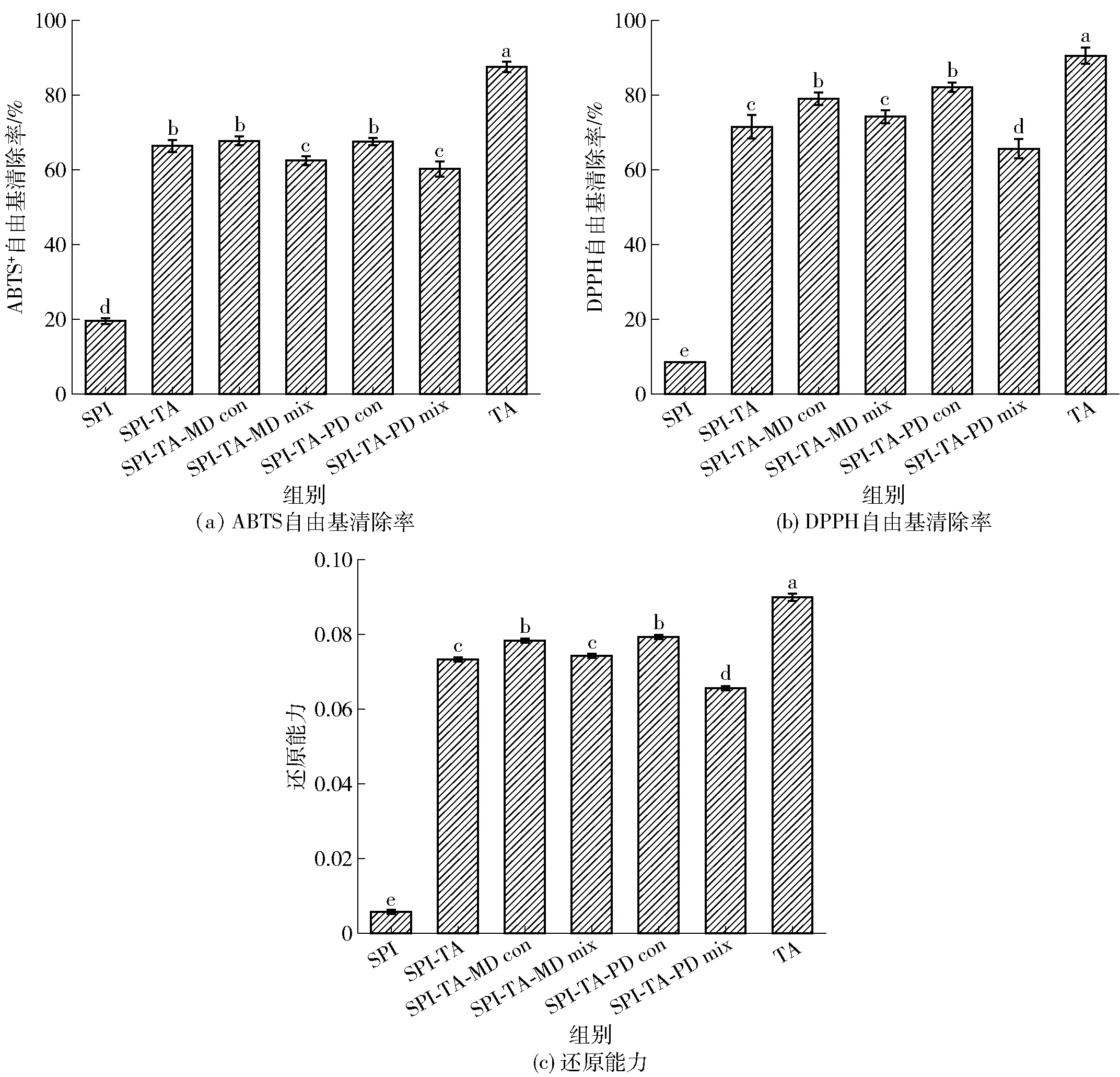
不同小写字母代表同一指标差异显著(P<0.05)。
2.7 共价接枝对SPI乳化活性和乳化稳定性的影响
与多糖发生美拉德反应是提升蛋白乳化活性的重要手段[54],多酚的共价结合同样会影响蛋白的乳化性能。SPI、SPI-TA、三元共价复合物和三元物理混合物的乳化活性、乳化稳定性如表2所示。
由表2可知,SPI在接枝TA、MD、PD后形成的共价复合物的乳化活性显著上升(P<0.05).相比于天然SPI,SPI-TA的乳化活性提高了149.04%。这是由于反应过程中TA的羟基与SPI的残基结合,导致蛋白质的结构改变,更多的疏水基团暴露在蛋同列不同小写字母表示组间差异显著(P<0.05)。
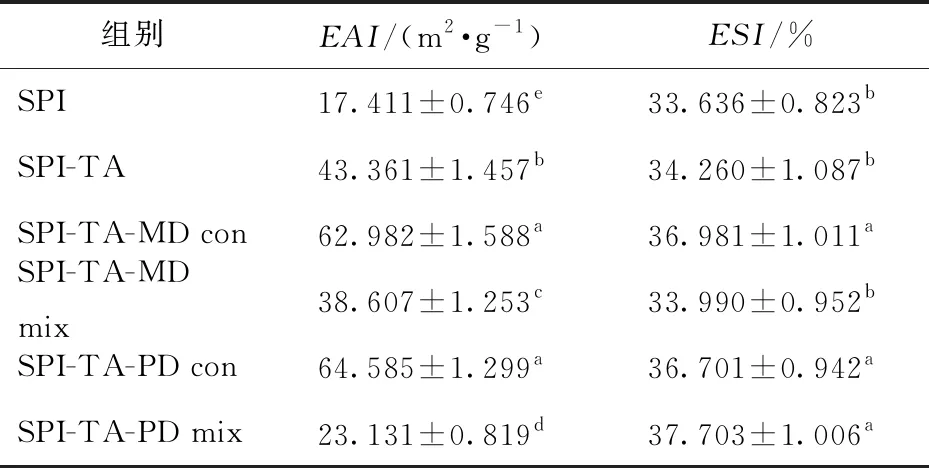
表2 SPI及其共价复合物和物理混合物的乳化活性和乳化稳定性
白表面,增加了SPI与油滴的结合能力,因此乳化能力提高。SPI-TA-MD、SPI-TA-PD共价复合物的乳化活性相比于SPI-TA分别提高了45.25%、48.95%,并且都显著高于其物理混合物(P<0.05)。这是因为湿法美拉德反应进一步打开了SPI的结构,增强了其在油水两相界面的吸附作用。此外,相比于SPI和SPI-TA,SPI-TA-MD、SPI-TA-PD共价复合物的乳化稳定性也显著提高(P<0.05),这是因为蛋白质表面吸附大量多糖后,蛋白质分子之间的强静电排斥会产生空间位阻,形成的乳液液滴较小,从而得到更加稳定的乳液[55]。
2.8 共价接枝对SPI起泡性和泡沫稳定性的影响
起泡特性是衡量许多食品品质的重要因素,比如牛奶、冰淇淋、鲜奶油、蛋糕和面包等[8]。蛋白质溶液经快速搅打使大量气体进入溶液中,降低其表面张力后形成泡沫的能力称作蛋白的起泡性[56],泡沫稳定性是指泡沫产生后稳定泡沫的能力,影响起泡能力的因素多且不易控制[28]。SPI及其共价复合物和物理混合物的起泡性、起泡稳定性如图7。
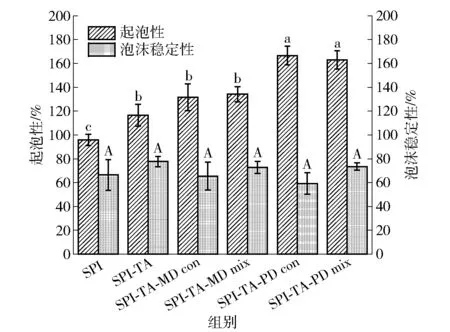
不同小写字母代表起泡性差异显著(P<0.05),不同大写字母代表起泡稳定性差异显著(P<0.05)。
由图7可知,SPI的起泡性为95.75%,SPI-TA的起泡性为116.44%,共价接枝了TA后,SPI的起泡性显著提高(P<0.05)。这是因为SPI和TA形成的共价交联结构可以使蛋白在气-液表面的快速吸附以及展开重排,增强了泡沫的形成能力[21]。多糖的加入同样提高了蛋白的起泡性,SPI-TA-PD共价复合物和物理混合物的起泡性能分别为166.67%、162.99%,相比于SPI-TA二元共价复合物分别提高了50.23%、46.55%。一方面是因为PD的引入显著提高了样品溶液的黏度,使得其起泡性显著提高(P<0.05);另一方面,SPI-TA-PD共价复合物中的蛋白具有较好的溶解性(图5),这允许蛋白质分子更有效地转移到空气- 水界面[57]。并且不同多糖的加入,起泡性的改变程度不同,SPI-TA-PD共价复合物的起泡性显著优于SPI-TA-MD共价复合物(P<0.05)。这和多糖的性质有关,PD的水溶液比MD的水溶液具有更高的黏度。剪切30 min后测得各个样品的泡沫稳定性没有显著差异,表明TA、MD、PD的加入不会对蛋白的泡沫稳定性产生不利影响。SPI-TA的泡沫稳定性略高于SPI,这是因为多酚的加入降低了蛋白界面的表面压力,使得界面薄膜变得更加稳定。
2.9 共价接枝对SPI表面疏水性的影响
表面疏水性是评价蛋白质理化性质的重要指标,与其乳化特性密切相关。SPI及其共价复合物的表面疏水性实验结果如图8。由图8可知,SPI在接枝TA、MD、PD后,共价复合物的表面疏水性显著提高(P<0.05)。SPI-TA表面疏水性提高是因为TA附着在亲水性的氨基酸上,并且共价接枝使得SPI的构象展开,导致内部疏水基团暴露在SPI分子表面上[58]。Mendis等[59]研究发现β-折叠含量的减少表明疏水相关位点可能暴露。研究结果中SPI-TA二级结构中β-折叠减少(图4)和疏水性的提高证实了这一点。SPI-TA-MD、SPI-TA-PD共价复合物的表面疏水性更高,这和湿热处理有关。热处理使蛋白质的疏水基团进一步暴露,二级结构的改变同样影响了三元共价复合物的表面疏水性。同干法糖基化相比,湿法美拉德反应可导致更多的疏水基团暴露于蛋白质表面[20]。本研究表明,TA、MD、PD的共价接枝可以改善SPI的表面疏水性。
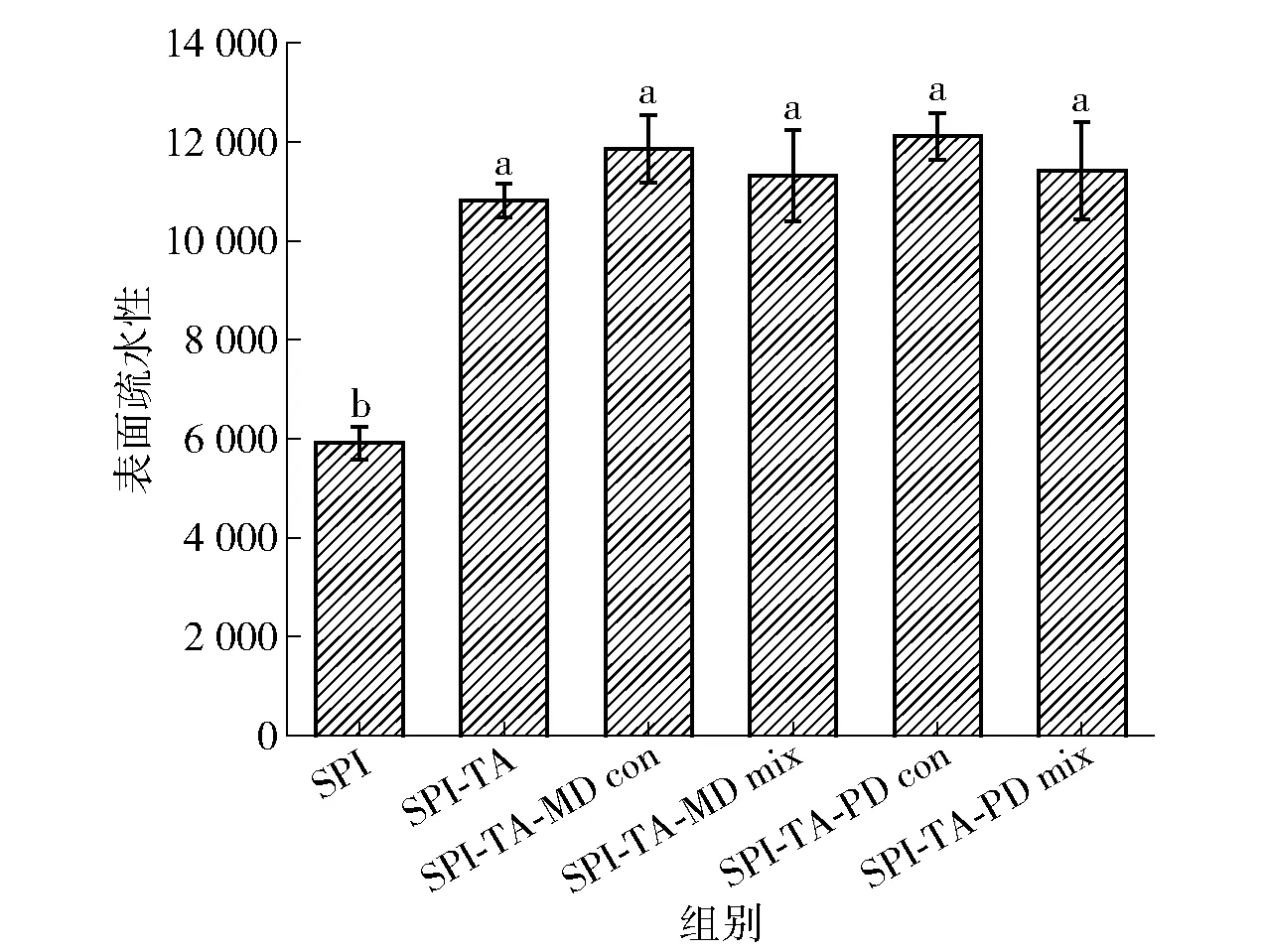
不同小写字母代表组间差异显著(P<0.05)。
2.10 共价接枝对SPI热稳定性的影响
加热可以改变蛋白质分子间的作用力,破坏其内部的化学键,变性温度越高,说明热稳定性越高。差示扫描量热(differential scanning calorimetry,DSC)是一种有效的热分析手段。样品的DSC图谱见图9。由图9可知,图中的吸热峰值主要归因于SPI的热变性,SPI-TA的变性温度为117.53 ℃,高于SPI的变性温度108.5 ℃,说明TA对SPI的共价修饰提高了SPI的热稳定性。这是因为SPI经过TA修饰之后,二级结构发生变化。Liu等[19]研究也发现乳铁蛋白与绿原酸共价后其变性温度提高了4.29 ℃。SPI-TA-MD、SPI-TA-PD物理混合物的变性温度分别为115.34、119.37 ℃,SPI-TA-MD、SPI-TA-PD共价复合物的变性温度分别为111.14、110.39 ℃。2种三元共价复合物的变性温度均高于天然SPI,但是低于SPI-TA及其对应的物理混合物。一方面归因于SPI-TA分子被MD、PD共价修饰后有序结构比例的降低以及无规则卷曲结构的增加,这和蛋白质二级结构的研究结果一致;另一方面,这可能是因为共价复合物在湿法美拉德反应条件下部分蛋白变性导致的。本研究结果表明,TA和MD、PD的共价接枝可以对SPI的热变性起到一定的保护作用。
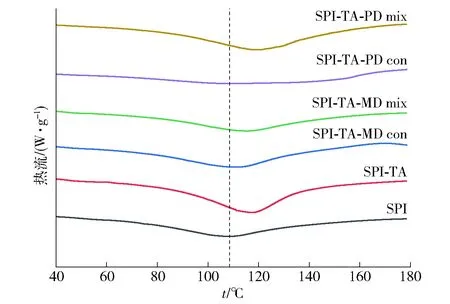
图9 SPI及其共价复合物和物理混合物的 差示扫描量热分析
3 结 论
以SPI、TA、MD、PD为原料,通过碱处理法、湿法美拉德反应制备SPI-TA共价复合物和SPI-TA-MD、SPI-TA-PD三元共价复合物,研究多酚(TA)和多糖(MD、PD)的共价修饰对SPI结构和功能特性的影响。通过反应基团的光谱以及电泳分析,证实SPI-TA和SPI-TA-MD、SPI-TA-PD共价复合物的形成。SPI-TA中TA的接枝量为(35.56±1.32) μmol/g,SPI-TA-MD、SPI-TA-PD三元共价复合物中MD和PD的接枝度分别为46.54%、32.26%。紫外- 可见光吸收光谱和红外光谱分析SPI及其共价复合物和物理混合物的结果表明:TA、MD、PD的共价接枝改变了SPI的结构,使SPI的β-折叠含量下降,α-螺旋、β-转角和无规则卷曲的含量增加;相比于SPI、SPI-TA和三元物理混合物,同时共价接枝了多酚和多糖的SPI-TA-MD、SPI-TA-PD三元共价复合物表现出更加优异的溶解性、乳化性、抗氧化活性、起泡性、表面疏水性和热稳定性。2种三元共价复合物具有较为相似的结构和功能特性,但是SPI-TA-PD共价复合物的起泡性显著优于SPI-TA-MD共价复合物。SPI-TA-MD、SPI-TA-PD三元共价复合物可以作为新型的乳化剂、抗氧化剂和起泡剂用于食品工业,以期为大豆分离蛋白的结构和功能特性优化提供新思路。
