基于房颤中circRNA-miRNA-mRNA 网络构建和免疫细胞浸润的生物信息学分析
2022-12-15范吉林朱婷婷田晓玲刘思佳张世亮
范吉林, 朱婷婷, 田晓玲, 刘思佳, 苏 静, 张世亮
(1. 山东中医药大学第一临床医学院, 山东 济南 250014;2. 滨州医学院附属医院神经外科, 山东 滨州 256600;3. 山东中医药大学附属医院心病科, 山东 济南 250014)
心房颤动(atrial fibrillation, AF)是以快速和无序的心房电活动为特征的室上性快速性心律失常, 是最常见的心律失常之一[1]。免疫细胞浸润在AF 的发展中有重要作用。研究[2]显示:巨噬细胞会导致AF 中的结构重塑。此外, AF 时M1 巨噬细胞极化的百分率明显升高, M1 巨噬细胞极化可有效促进心房纤维化, 而M2 巨噬细胞极化可改善心房纤维化[3]。此外, 有研究[4]显示:单核细胞中增强的迁移活性可能在AF 患者心房重塑进展中起关键作用。然而, 目前关于CD4 T 淋巴细胞在AF发病机制中的具体作用机制尚不清楚。因此, 有必要进一步深入了解AF 免疫细胞浸润的机制, 开发出更有效的AF 治疗方法。
环状RNA (circular RNA, circRNA) 是一种在特定生物过程中具有调节功能的新型非编码RNA[5], 其 能 够 与 内 源 性RNA (competitive endogenous RNA, ceRNA) 竞争性抑制特定微小RNA(microRNA, miRNA)的活性, 从而调节基因表达并充当miRNA 的“海绵”[6]。目前已知circRNA 参与动脉粥样硬化[7]、心肌梗死[8]和心力衰竭[9]等心血管疾病的发生发展。此外, 研究[10]显示:circRNA 可能通过序列驱动的海绵效应与miRNA 相 互 作 用, circRNA- miRNA-mRNA 网 络在心血管疾病的生理和病理过程中均发挥作用。WU 等[11]研 究 显 示:mmu_circ_0005019 可 以 通 过充当miR-499-5p 海绵来调节其靶基因Kcnn3 的表达, 从而在AF 中抑制心脏成纤维细胞的纤维化并逆转心肌细胞的电重构。因此, 通过构建circRNAmiRNA-mRNA 调控网络可以为AF 的病理生理学研究提供新的路径。然而, 目前对AF 中免疫细胞浸润以及免疫相关circRNA-miRNA-mRNA 网络之间关系的研究较少。
本研究采用基因表达数据库 (Gene Expression Omnibus, GEO) 数据库和生物信息学方法, 挖掘AF 和正常组织中显著差异表达的circRNA、miRNA 和mRNA, 构建circRNA 相 关的ceRNA 调控网络。此外, 采用CIBERSORT 软件在AF 中进行免疫细胞浸润分析, 采用Pearson相关系数法分析与AF 中免疫细胞浸润相关的基因, 旨在为circRNA 在AF 发展中的作用及其潜在作用机制并优化AF 治疗方案研究提供依据。
1 资料与方法
1.1 数据集获取和差异表达分析从基因表达综合数据库中筛选并获取AF 和相应正常组织的微阵列circRNA (GSE129409)、miRNA (GSE28594)和mRNA(GSE41177)的基因表达谱数据。采用R 软件中的“limma”数据包来确定每个数据集中的DEmRNA、DEmiRNA 和DEcircRNA, 标准为|log2(FC) |>1 且P<0.05[12]。采用R 软件中的“ggplot2”和“pheatmap”数据包绘制差异表达基因(differential expression genes, DEGs)的火山图和聚类图。
1.2 circRNA-miRNA-mRNA 网络构建circRNA和miRNA 之间的相互作用在疾病调控中起重要作用[13]。首先采用ENCORI[14]和circBank[15]数据库来预测与circRNA 结合的miRNA。相应地, 数据库中预测的miRNA 与R 软件中分析得到的差异表达 miRNA (differentially expressed miRNA, DEmiRNA) 的 交 集 视 为 关 键miRNA。 采 用TargetScan[16]和miRDB[17]数据库预测与miRNA结合的mRNA, 并与R 软件中分析得到的DEmRNA 取交集视为关键mRNA。最后, 将所有结果导入Cytoscape3.7.2 软件构建circRNAmiRNA-mRNA 网络。
1.3 基因本体论(Gene Ontology, GO)和京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)富集分析DAVID 数据库是一个基因在线注释工具网站, 将circRNAmiRNA-mRNA 网络中的DEmRNA 列表导入DAVID 数据库中, 得到这些基因的GO 和KEGG富集分析结果[18]。
1.4 免疫细胞浸润分析采用CIBERSORT[19]软件计算每个样本中22 种免疫细胞浸润的百分率, 将P<0.05 的样本输出用于后续分析。采用R 软件对22 种免疫细胞浸润性进行Spearman 相关分析, 并采用“corrplot” 数据包绘制相关热图以可视化结果。同时采用R 软件中的“vioplot”数据包比较和可视化AF 与正常样本之间的22 个免疫细胞浸润百分率。
1.5 ceRNA 调控网络中关键标志物的鉴定采用受试者工作特征(receiver operating characteristic, ROC)曲线确定circRNA-miRNA-mRNA 调控网络中关键标志物的诊断价值。ROC 曲线下面积(area under curve, AUC) 大 于 0.8 的 关 键circRNA、miRNA 和mRNA 被确定为AF 中的潜在诊断生物标志物。
1.6 关键mRNA 与免疫细胞浸润的相关性分析
采用Pearson 相关系数法分析关键mRNA 与免疫细胞浸润之间的相关性, 并采用R 软件中的“ggpubr”数据包进行可视化。
1.7 统计学分析采用R 软件(版本4.0.5)进行统计学分析。对mRNAs 进行富集分析, 并采用CIBERSORT 软件计算22 种免疫细胞浸润百分率;采用ROC 曲线确定ceRNA 网络中关键标志物的诊断价值;采用Pearson 相关分析法分析关键mRNA与免疫细胞浸润之间的相关性。以P<0.05 为差异有统计学意义。
2 结 果
2.1 健 康 对 照 者 和 AF 患 者 DEcircRNA、DEmiRNA 和DEGs 鉴定与健康对照者的正常样本比较, GSE129409 共鉴定出AF 患者的492 个差异DEcircRNAs, 其中包括316 个上调circRNAs 和176 个下调的circRNAs, GSE41177 共鉴定出AF患者的296 个DEGs, 其中包括165 个上调基因和131 个下调基因, GSE28594 共鉴定出AF 患者的37 个DEmiRNAs, 其 中 包 括 14 个 上 调miRNAs 和23 个下调miRNAs。见图1。
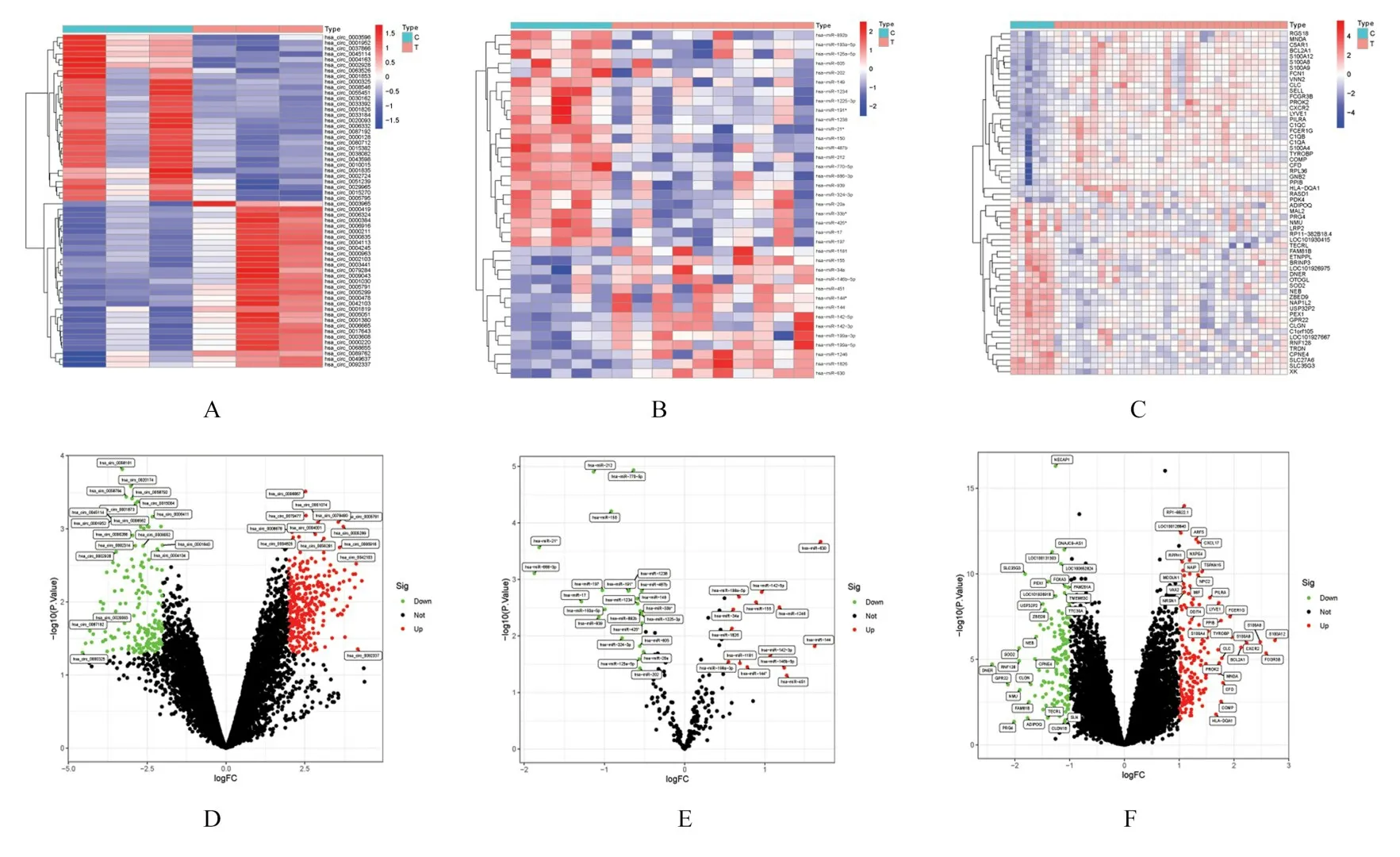
图1 DEGs 的聚类图(A-C)和火山图(D-F)Fig.1 Cluster maps(A-C)and bolcanic maps (D-F)of DEGs
2.2 ceRNA 调控网络的构建采用不同数据库对DEmiRNA、DEcircRNA 和DEmiRNA 之间的相互作用进行预测, 构建ceRNA 调控网络。预测出与DEcircRNA 相结合的miRNA 589 个, 将预测得到的miRNA 与DEmiRNA 取交集获得9 个miRNAs, 预测出DEmiRNA 的靶基因3 000 个, 将预测得到的靶基因与DEmiRNA 取交集获得32 个差异基因(图2)。最终建立1 个包含31 个基因(7 个circRNAs、 5 个miRNAs 和19 个mRNAs) 的ceRNA 调控网络。 这个调控网络包括7 个circRNA-miRNAs 对 和19 个miRNA-mRNAs 对。见图3 和表1。
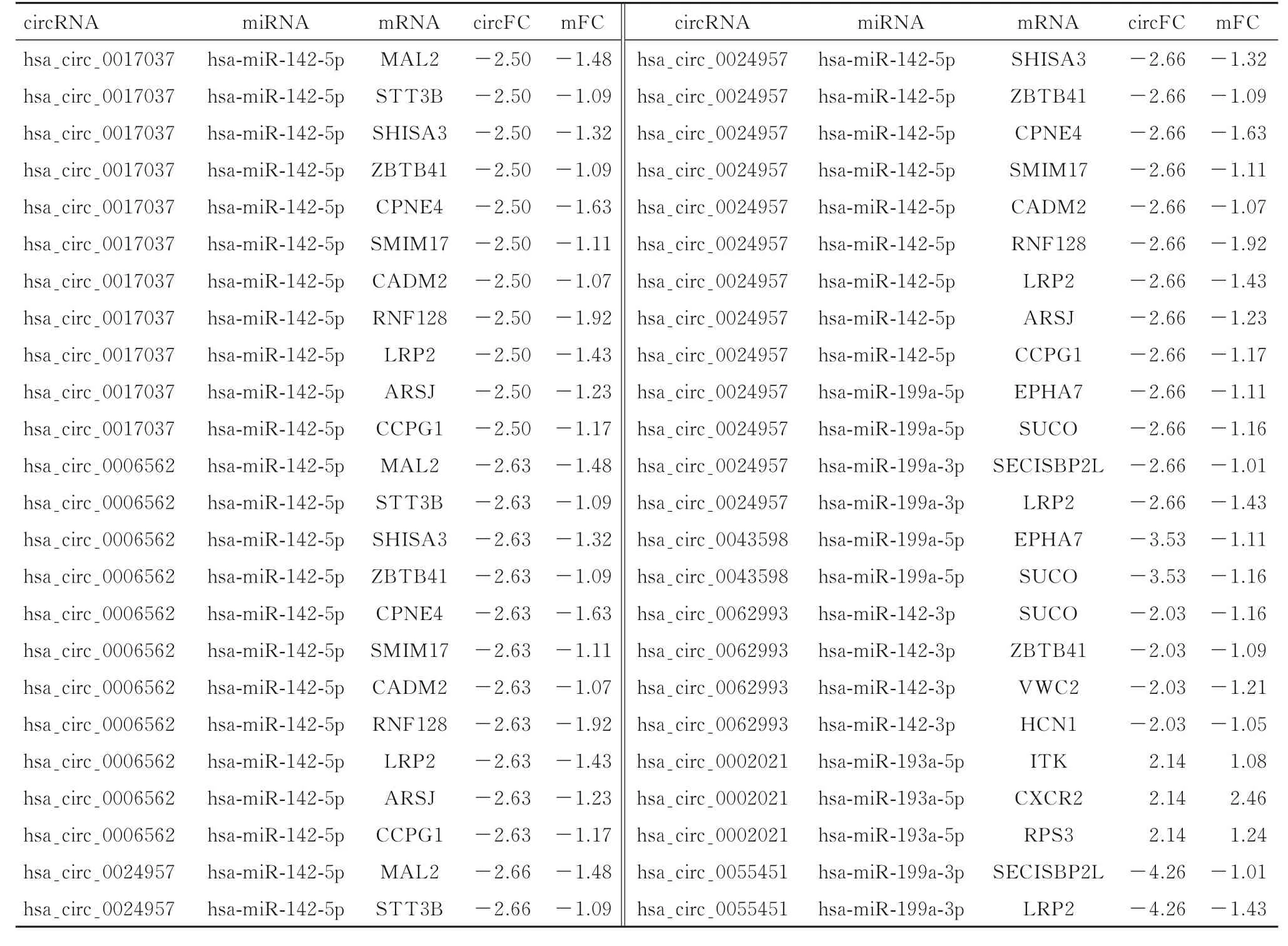
表1 ceRNA 网络中circRNA、miRNA 和mRNA 及其靶基因相互作用Tab.1 Interaction among circRNA, miRNA, mRNA and their target genes in ceRNA network
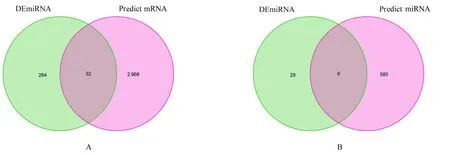
图2 DEmiRNA 与预测mRNA(A)和DEmiRNA 与预测miRNA(B)取交集的Venn 图Fig.2 Venn diagrams of intersections of DEmiRNA and predict mRNA(A)and DEmiRNA and predict miRNA(B)
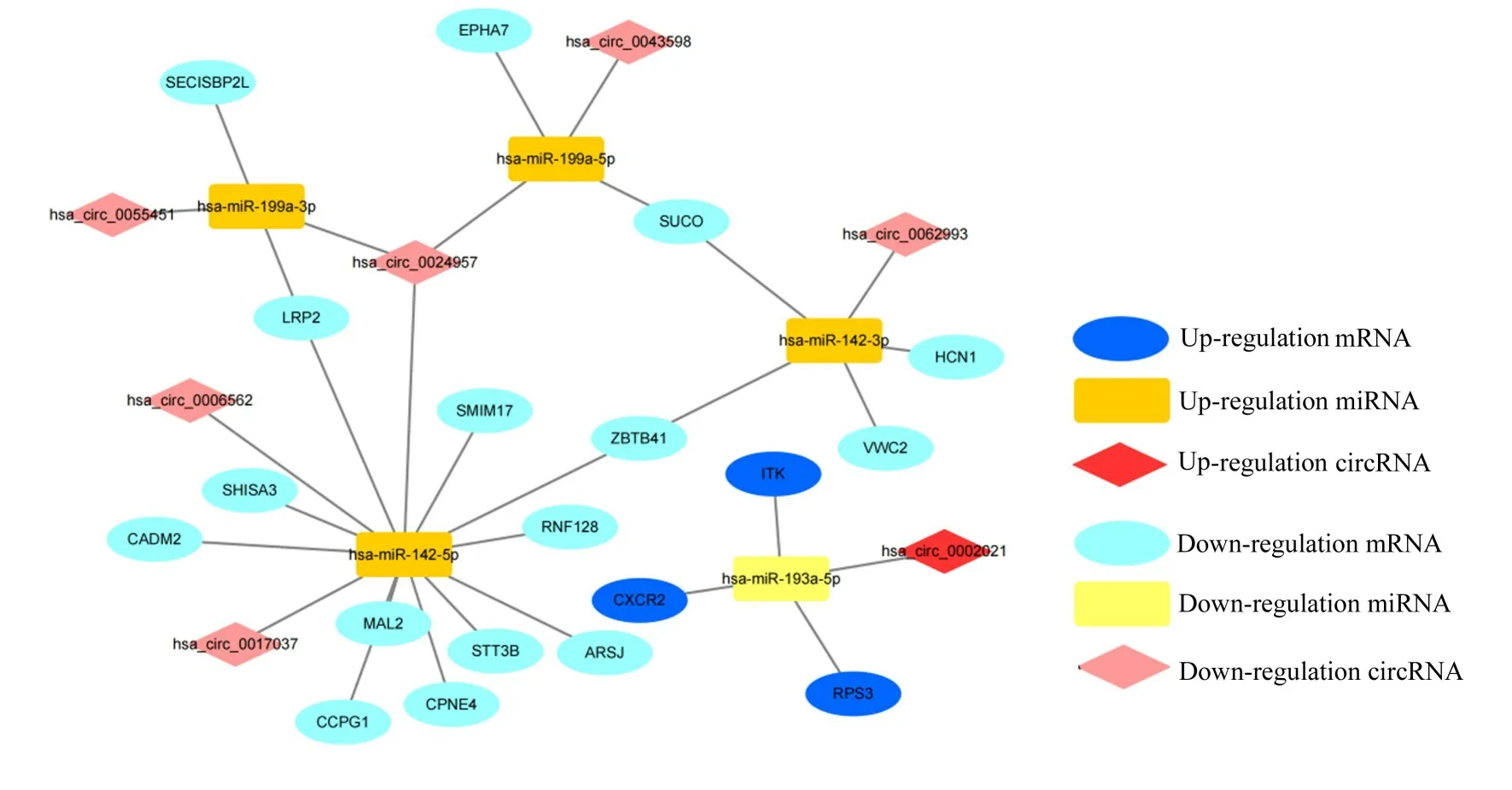
图3 AF 中circRNA-miRNA-mRNA 调控网络Fig.3 circRNA-miRNA-mRNA regulation network in AF
2.3 GO 和KEGG 富集分析对ceRNA 调控网络中的DEmRNA 进行GO 和KEGG 富集分析。GO富集分析结果显示:这些基因在蛋白质降解、细胞的胞吐作用、蛋白酪氨酸激酶活性和环核苷酸门控离子通道活性等方面显著富集。KEGG 富集分析结果表明:这些基因在趋化因子信号通路、刺猬信号通路、T 细胞受体信号通路和细胞-细胞因子相互作用等信号通路中显著富集。见图4。
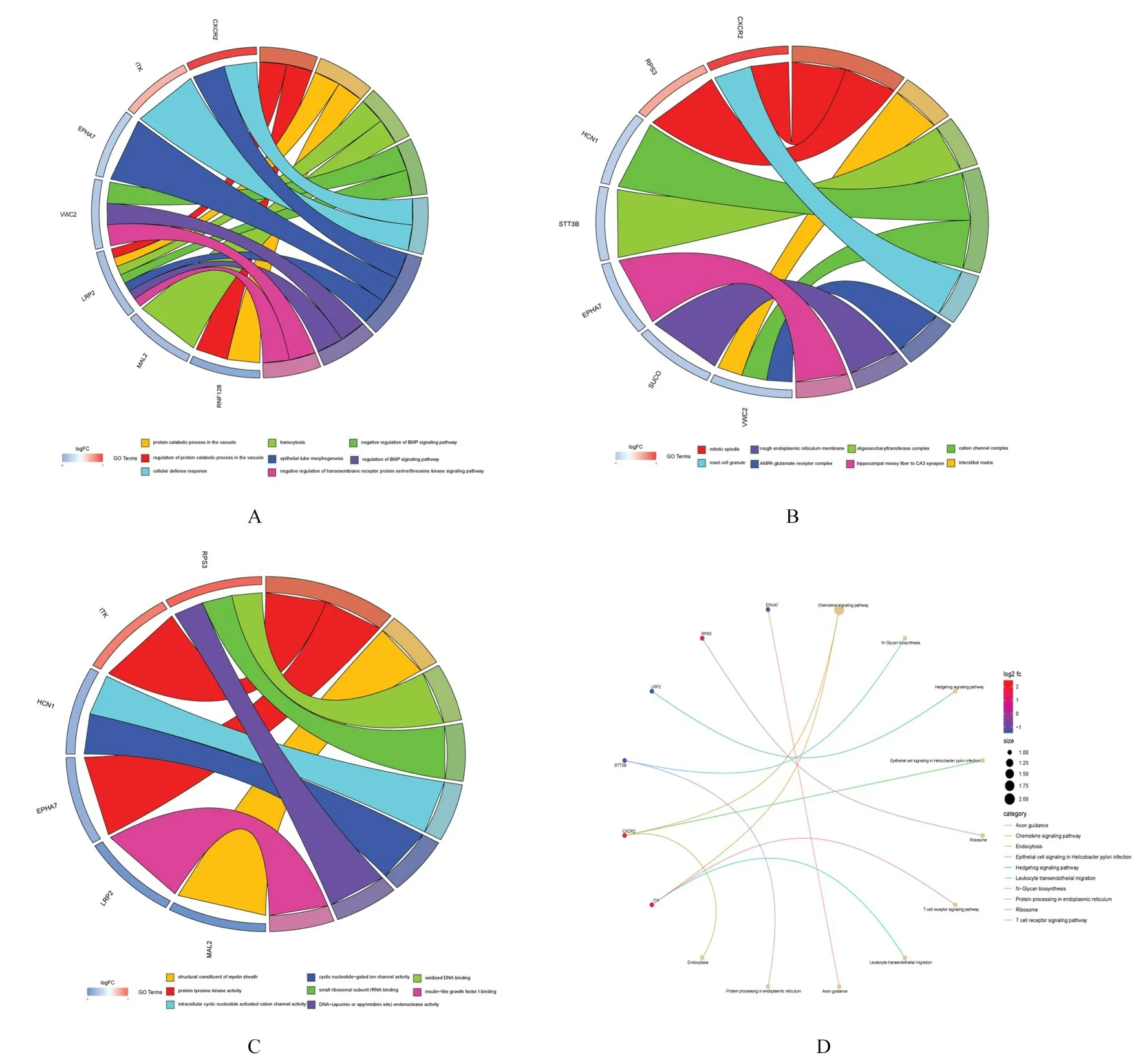
图4 GO 和KEGG 富集分析图Fig.4 Diagrams of GO and KEGG enrichment analysis
2.4 mRNA 数据集中的免疫浸润百分率首先分析每个样品中免疫细胞的组成并通过条形图显示其组成(图5A);分析数据集中免疫细胞表达的相关性的结果显示:嗜酸性粒细胞和静息CD4+记忆T 淋巴细胞与M1 巨噬细胞表达水平、静息CD4+记忆T 淋巴细胞与γδT 淋巴细胞表达水平、中性粒细胞与单核细胞表达水平、M1 巨噬细胞与γδT 淋巴细胞表达水平、滤泡辅助T 淋巴细胞与活化的树突状细胞表达水平呈正相关关系;中性粒细胞与M2 巨噬细胞表达水平, CD8+T 淋巴细胞表达水平、静息CD4+记忆T 淋巴细胞与单核细胞表达水平呈负相关关系(图5B);分析AF 样本和正常样本之间免疫细胞表达的差异, 并采用小提琴图进行可视化显示:在正常样本中, 滤泡辅助T 淋巴细胞和活化的树突状细胞浸润百分率较高(P<0.05), 而在AF 样本中, γδT 淋巴 细胞、单核细胞、M2 巨噬细胞和中性粒细胞浸润百分率较高(P<0.05)(图5C)。
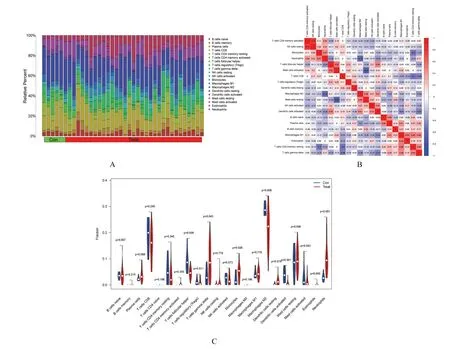
图5 mRNA 免疫浸润分析Fig.5 Analysis on mRNA immune infiltration
2.5 ceRNA 调控网络中关键标志物的ROC 曲线分析ROC 曲线分析结果显示:MAL2、STT3B、SHISA3、 ZBTB41、 CPNE4、 EPHA7、 SUCO、SMIM17、VWC2、ITK、CXCR2、SECISBP2L、HCN1、 CADM2、 RNF128、 LRP2、 RPS3、ARSJ 和 CCPG1 等 mRNA, hsa_circ_0017037、hsa_circ_0006562、 hsa_circ_0024957、 hsa_circ_0043598、 hsa_circ_0062993、 hsa_circ_0002021、hsa_circ_0055451 等 circRNA 和 hsa-miR-142-5p、hsa-miR-199a-5p、hsa-miR-142-3p、hsa-miR-193a-5p、hsa-miR-199a-3p 等miRNA AUC 均在0.8 以上, 表明以上调控网络具有较高的预测价值。见图6。
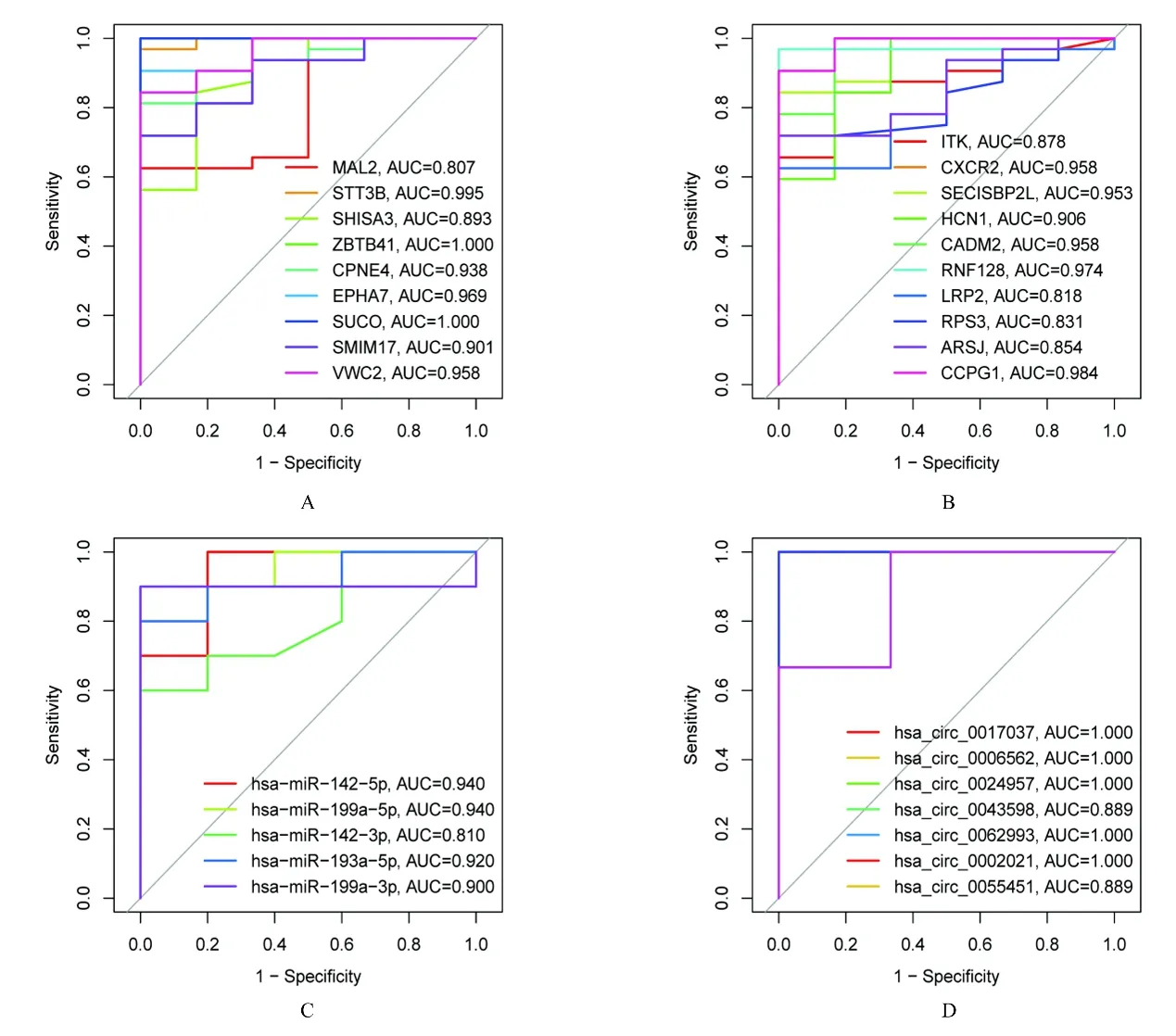
图6 关键mRNAs、miRNAs 和circRNAs 的ROC 曲线Fig.6 ROC curves of key mRNAs, miRNAs, and circRNAs
2.6 关键mRNA 与免疫细胞浸润的关系通过对ceRNA 调控网络中的19 个mRNA 与22 个免疫细胞浸润的相关性分析, 发现CXCR2 与中性粒细胞(r=0.85,P<0.001)、单核细胞(r=0.32,P=0.04)和γδT 淋巴细胞(r=0.38,P=0.01)浸润呈正相关关系, 与M2 巨噬细胞(r=-0.66,P<0.001)、滤泡辅助T 淋巴细胞(r=-0.38,P=0.02)浸润呈负相关关系;CADM2 与M2 巨噬细胞(r=0.45,P=0.004)、活化的树突状细胞(r=0.45,P=0.004) 和 静 息 肥 大 细 胞(r=0.43,P=0.007) 浸润呈正相关关系, 与浆细胞(r=-0.33,P=0.03)、单核细胞(r=-0.37,P=0.01) 和中性粒细胞(r=-0.45,P=0.004) 浸润呈负相关关系。见图7。
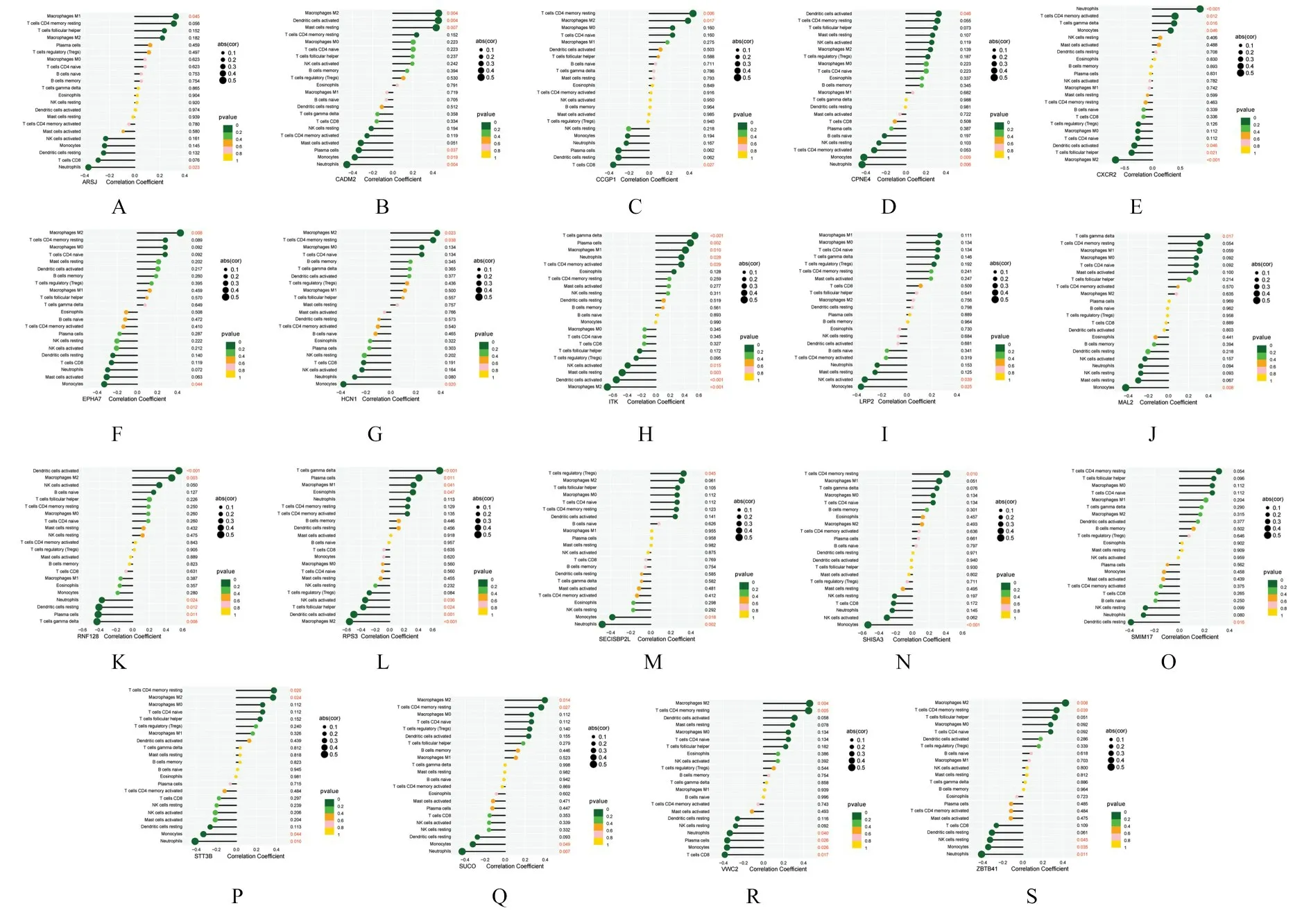
图7 mRNA 与免疫细胞浸润的相关性Fig.7 Correlation between mRNA and immune cells infiltration
3 讨 论
AF 不仅给患者个人带来巨大的身心痛苦, 也给国家带来沉重负担, 因此了解AF 的分子机制对于其临床诊治至关重要。高效的高通量测序和生物信息学分析有助于了解疾病发生发展的分子机制, 这对于探索遗传改变和识别潜在的诊断生物标志物十分必要。近年来, 研究重点已转向AF 的机制研究, 如mRNA、miRNAs 和circRNAs, 均在调节AF 中发挥不同作用。因此, 本文作者采用生物信息学方法分析了3 组AF 患者和正常对照者的微阵列数据, 包括mRNA、miRNA 和circRNA 数据集。
本研究中构建的ceRNA 网络包括19 个mRNAs、5 个miRNAs 和7 个circRNAs。circRNA
是一类新的ceRNA, 可以作为miRNA 的分子海绵, 抑制miRNA 的所有靶基因, 且已被证明在AF中发挥重要的调节作用[20]。在本研究构建的ceRNA 网络中, hsa_circ_0002021 与CXCR2 彼此竞争, 影响miR-193a-5p 的表达, CXCR2 的激活会诱发几个下游信号通路的激活, 包括核因子κB(nuclear factor kappa B, NF-κB)、NADPH 氧化酶和转化生长因子β1 (transforming growth factorbeta 1, TGF-β1)/Smad2/3, 促进促炎和促氧化反应, 从而导致AF 的纤维化[21], 因此, 阻断CXCR2 显著阻止和减少了这些有害影响;miR-193a-5p 及其家族miR-193 已被研究[22]确定与心血管疾病的发生发展有密切关联, 且miR-193a-5p 是多种肿瘤疾病的重要靶点, miR-193a-5p 的过表达或低表达有助于肿瘤细胞增殖、侵袭、迁移、凋亡和转移[23], 然而关于miR-193a-5p 与AF 的直接研究较少, 尚需进一步研究。miR-142-5p 的过表达通过下调SIRT7 导致缺氧暴露的H9C2 心肌细胞和原代心肌细胞的广泛细胞损伤及凋亡[24];WANG 等[25]研究显示:miR-142-5p 低表达与慢性心力衰竭显著相关。miR-142-5p 在单侧输尿管梗阻引起的肾纤维化中显著上调[26], 与本研究中miR-142-5p 表达上调趋势一致, miR-142-5p 表达的上调可能导致心房纤维化从而导致AF 的发生。hsa_circ_0006562、hsa_circ_0017037 和hsa_circ_0024957 的相关研究较少, 因此推测上述3 种circRNA 可能通过与miR-142-5p 竞争性结合来调控相关基因的表达, 参与AF 的发展。研究[27]显示:与健康受试者比较, 2 型糖尿病患者的miR-199a-3p 表达水平降低, 其机制可能与血管内皮细胞损伤有关, miR-199a-3p过表达会抑制血管内皮细胞的凋亡。本研究中miR-199a-3p 表达上调, 因此推测hsa_circ_0055451和hsa_circ_0024957 可能与miR-199a-3p 竞争性结合调控相关因子的表达, 从而改善AF 患者的内皮损伤, 预防并发症的发生。因此, 本文作者发现的circRNA 介导的ceRNA 调控网络可能为理解AF 发生发展的潜在机制提供新的见解。
为了进一步探讨AF 中差异表达mRNA 的生物学功能, 本文作者对ceRNA 网络中的19 个差异表达mRNA 进行了GO 和KEGG 富集分析。GO 富集分析结果显示:这些基因在蛋白质降解、细胞的胞吐作用、蛋白酪氨酸激酶活性和环核苷酸门控离子通道活性等方面显著富集。这与之前的研究[28]结论一致, 即AF 伴随着几种细胞变化, 例如心肌细胞凋亡和坏死、促炎介质的积累以及以纤维化诱导为特征的细胞外基质重组。KEGG 富集分析结果显示:mRNA 在趋化因子信号通路、刺猬信号通路、T 细胞受体信号通路、细胞-细胞因子相互作用和胆固醇代谢等信号通路中显著富集。趋化因子可分为不同亚家族, 即CC、CXC、CX3C 和XC, 趋化因子信号由其同源受体介导, 这些受体通常是与G 蛋白偶联的[29]。相关研究[30]显示:拮抗CXCR4可以防止AF 发展, 表明CXCL12/CXCR4 轴可能是AF 的潜在治疗靶点。另有研究[21]显示:在AF患者的心房组织中CXCL1 及其受体CXCR2 表达增加, 表明CXCR2 可以预防和逆转AF 的发展, 且CXCR2 可能是高血压患者AF 的潜在治疗靶点。另外富集得到的T 淋巴细胞受体信号通路和刺猬信号通路与AF 关系研究较少, 需通过实验进一步验证。
采用CIBERSORT 软件可以评估22 种免疫细胞的表达水平和动态调节过程。研究[2]显示:免疫反应与AF 之间的关系密切, 本研究结果显示:滤泡辅助T 淋巴细胞和活化的树突状细胞在正常样本中浸润百分率较高, 而γδT 细胞、单核细胞、M2 巨噬细胞和中性粒细胞在AF 样本中浸润百分率较高。研究[2]显示:巨噬细胞导致AF 患者的心房重塑, 与本研究中AF 样本中M2 巨噬细胞浸润百分率较高的结论一致。单核细胞迁移活性增强和心房壁单核细胞数量增加可能是心房重塑和AF 病理生理学的基础[4], 本研究中单核细胞在AF 组织中浸润百分率较高, 进一步说明生物信息学研究方法的合理性。中性粒细胞是关键的免疫炎症细胞[31], 高中性粒细胞与淋巴细胞百分率与新发和复发性AF 以及血栓栓塞性中风存在关联[32];研究[33]显示:中性粒细胞水平与AF 患者导管消融后的AF 复发呈正相关关系。目前关于γδT 淋巴细胞表达与AF 关系的相关研究较少, 需进一步完善相关实验, 探索其在AF 发生发展中的生物学机制。本研究分析和鉴定了正常组织和AF 组织中的差异表达circRNA, 并采用生物信息学方法构建了由7 个circRNAs、5 个miRNAs 和19 个mRNAs 组成的ceRNA 调控网络。此外, 鉴定出的关键mRNA 与AF 中免疫细胞的浸润呈相关关系。本研究结果为AF 中ceRNA 调控网络的潜在机制研究提供了新的见解, 同时也为寻找预防和治疗AF 的新靶点提供理论依据, 然而, 本研究得出的结论需要实验进一步验证。
