草酸艾司西酞普兰片粉末直压制备及质量研究
2022-12-12王丽春魏鸿波陆纪宏
林 琳 张 涛 王丽春 魏鸿波 陆纪宏
浙江金华康恩贝生物制药有限公司,浙江金华 321016
草酸艾司西酞普兰[1-3]是由美国Forest Lab 公司与丹麦H.Lundbeck A/S 公司于1997年开始联合开发,于2002年获美国食品药品监督管理局(FDA)批准,商品名为LEXAPRO®,草酸艾司西酞普兰是西酞普兰的S-对应异构体,是一种选择性的5-羟色胺再摄取抑制剂。适用于成年及12~17 岁青少年重度抑郁症(MDD)患者的急性和维持治疗和成年患者的广泛性焦虑症(GAD)的急性治疗,具有良好的耐受性和服药依从性。目前已上市的剂型包括片剂(5、10、20 mg)、口服溶液和胶囊。本研究以H.Lundbeck A/S 公司生产的草酸艾司西酞普兰片作为参比制剂,采用粉末直接压片的方式进行产品制备,采用正交试验法确定最佳处方,对自制制剂和参比制剂进行体外溶出比较,同时对参比制剂和自制制剂开展加速稳定性考察,比较自制制剂与参比制剂质量。
1 仪器与试剂
1.1 仪器
三维运动混合机(江苏金斯瑞机械设备有限公司),2020 压片机(菲特),XPE105 分析天平(梅特勒-托利多有限公司),FE28 型pH 计(梅特勒-托利多仪器有限公司),Acquity Arc 2998PDA 高效液相色谱仪(Waters),KQ800KDE 超声波清洗器(广东固特超声股份有限公司),SNTR-8400A 智能溶出仪(SHIMADZU Excellence in Science),UN110plus自然对流烘箱(memment),KBF115 恒温恒湿箱(德国宾德),KBF720 恒温恒湿箱(德国宾德),FT2000A 颗粒和粉末特性分析仪(瑞柯仪器有限公司)。
1.2 试剂
草酸艾司西酞普兰原料药(浙江海森药业有限公司,纯度质量分数≥99.0%),草酸艾司西酞普兰对照品(浙江海森药业有限公司,纯度质量分数≥99.7%),草酸艾司西酞普兰杂质对照品(USP),市售草酸艾司西酞普兰片(H.Lundbeck A/S,商品名:LEXAPRO®),微晶纤维素(Asahi Kasei Corporation;Asahi Kasei Corporation)、交联羧甲基纤维素钠(林州市环宇药用辅料厂)、胶态二氧化硅(美国卡博特)、硬脂酸镁(长葛市白茹药用材料有限公司)、滑石粉(广西龙胜华美滑石粉开发有限公司)、薄膜包衣预混剂(胃溶型)85G68918-CN[赢创特种化学(上海)有限公司],乙腈(色谱纯,山东鑫赢舜新材料有限公司),磷酸(色谱纯,TEDIA),盐酸(色谱纯,赛默飞),磷酸二氢钾(色谱纯,吴江市永和精细化工有限公司),氢氧化钠(色谱纯,国药集团化学试剂有限公司),冰醋酸(色谱纯,赛默飞),醋酸钠(色谱纯,国药集团化学试剂有限公司)。
2 方法与结果
2.1 草酸艾司西酞普兰片样品的制备
称取处方量的原料及辅料,将草酸艾司西酞普兰与胶态二氧化硅先进行混合5 min[4],过40 目筛,然后将该混合物再加入三维运动混合机中,加入微晶纤维素PH102、交联羧甲基纤维素钠、硬脂酸镁、滑石粉,混合15 min,用直径为7.0 mm 的冲模,直接压片[5-6],再采用薄膜包衣预混剂进行包薄膜衣。
2.2 质量评价指标与方法
2.2.1 休止角 采用粉体流动性FT2000A 颗粒和粉末特性分析仪检测中间体草酸艾司西酞普兰片颗粒的休止角,每批样品各检测3 次,取其平均值。
2.2.2 脆碎度 按照《中华人民共和国药典2020年版》(4 部)通则0923 片剂脆碎度检查法测定,减失重量不得过1%,并不得出现断裂、龟裂及粉碎的片[7]。
2.2.3 溶出度 按照《中华人民共和国药典2020年版》(2 部)草酸艾司西酞普兰片质量标准检测[7]。
2.3 处方优化与正交试验设计
2.3.1 因素与水平[8-9]本品制备工艺为粉末直压法,主要考察最终混合后的物料流动性是否满足生产需求,压片后片子的脆碎度和溶出度需符合标准要求。本品采用原料与辅料微晶纤维素和交联羧甲基纤维素钠按既定比例混合后,直接压片,可制得片子硬度和溶出度均符合质量要求的产品。为确定各辅料的最终用量,本试验计划选择表1中A、B、C 三个因素为影响因素,并分别取3 个水平,以片剂脆碎度、休止角、溶出度三个指标,进行处方筛选优化。因素与水平见表1。

表1 正交试验因素与水平表
2.3.2 综合评分方法[10]总混后测定物料的休止角(a)、压片后测定片子的脆碎度(b)及第30 分钟测定的溶出度(c),通过上述三个指标进行综合评估。休止角反映颗粒的流动性,脆碎度反映物料的可压性,正交试验综合评分公式为:M=(50-a)×2+(1-b)×20+c×0.4。
2.3.3 正交试验结果 通过正交试验结果分析可知,胶态二氧化硅比例(B)影响显著,交联羧甲基纤维素钠比例(A)和硬脂酸镁比例(C)影响不显著。影响因素的优先顺序为B>C>A,最佳制备处方为交联羧甲基纤维素钠所占比例为5.0%,胶态二氧化硅为2.0%,硬脂酸镁为1.0%。见表2。

表2 正交试验结果
2.4 优选处方验证
依照优选的处方工艺制备三批草酸艾司西酞普兰片样品。按照《中华人民共和国药典2020年版》(2 部)草酸艾司西酞普兰片质量标准进行检测[7],并与参比制剂开展体外溶出行为考察,分别取一批自制制剂和一批参比制剂进行加速稳定性考察,以进一步验证处方工艺的可行性。
2.5 质量检测
2.5.1 外观 所制得的片剂为薄膜衣片,除去包衣后显白色。符合《中华人民共和国药典2020年版》(2 部)草酸艾司西酞普兰片质量标准[7]。
2.5.2 有关物质 按照《中华人民共和国药典2020年版》(2 部)草酸艾司西酞普兰片质量标准检测,结果表明,自制制剂有关物质均符合标准要求[7]。见表3。
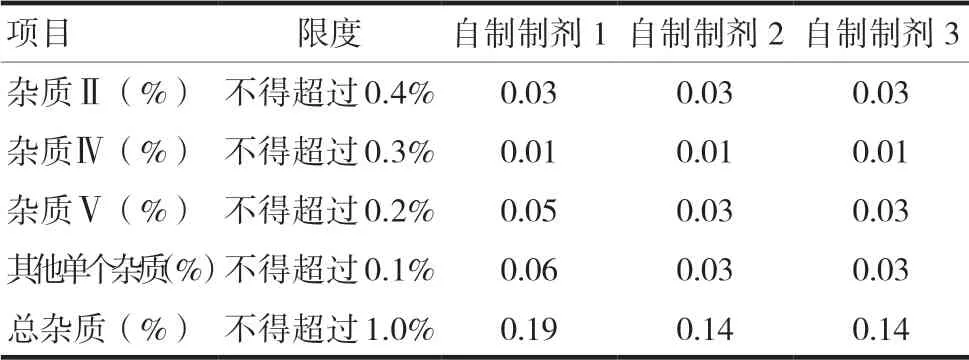
表3 自制制剂有关物质考察结果
2.5.3 含量及含量均匀度 按照《中华人民共和国药典2020年版》(2 部)草酸艾司西酞普兰片质量标准检测[7],三批仿制制剂含量分别为101.30%、100.30%、100.40%(限度为95.00%~105.00%),含量均匀度分别为4.90、2.80、3.40(限度初试:≤15.00)。结果表明,自制制剂含量和含量均匀度均符合标准要求。
2.5.4 参比制剂与自制制剂溶出曲线比较与分析[11-12]取本品,照溶出度与释放度测定法(通则0931 第二法)。溶出介质:水、pH 1.0 盐酸溶液、pH 4.5 醋酸溶液及pH 6.8 磷酸盐缓冲液;转速:50 r/s;体积:900 ml;取样时间:分别于5、10、15、30 min 取样。测定法:照《中华人民共和国药典2020年版》(2 部)草酸艾司西酞普兰片质量标准含量检测项的色谱条件,量取对照品溶液与供试品溶液各20 μl,注入液相色谱仪,按外标法以峰面积计算每片的溶出量,原研制剂和自研产品在不同介质中的溶出曲线见图1~4。参比制剂与自制制剂在溶出介质pH 1.0、pH 4.5、pH 6.8 中,取样点15 min 时,溶出度均达85%以上,按照《普通口服固体制剂溶出曲线测定与比较指导原则》[13]规定:“当受试制剂和参比制剂在15 min 内的溶出量≥85%时,可以认为两者溶出行为相似,无需进行f2 的比较。”在水介质中,三批自研产品与原研制剂进行f2 值计算分别为59.60、68.50、62.00,结果均大于50,即自研产品与原研制剂在介质水中的溶出曲线具有相似性。可确定自研产品与原研产品体外溶出行为一致[14-15]。
2.6 稳定性考察
将自制制剂按市售包装与参比制剂置于40℃±2℃,相对湿度75%±5%条件下考察6 个月,分别于0、1、2、3、6 个月测定含量、溶出度和有关物质与参比制剂进行比较。结果表明,自制制剂与仿制制剂放置6月后各项检测指标均无明显变化。由此可见,粉末直压法制备的草酸艾司西酞普兰片剂含量、含量均匀度、溶出度、有关物质均无明显变化,质量稳定。见表4。

表4 稳定性试验数据
3 讨论
草酸艾司西酞普兰对湿热极其不稳定,采用常规方法湿法制粒,极易导致发生水解等反应有关物质上升,导致疗效降低,本研究采用粉末直压工艺,降低了产品质量风险,缩短了生产周期,降低生产成本,所制备的草酸艾司西酞普兰片经加速6 个月的留样考察,质量结果与参比制剂相似,产品质量稳定,达到预期目的。
