新型HIV-1附着抑制剂fostemsavir
2022-12-08王珺,展鹏
王 珺, 展 鹏
艾滋病(AIDS)是由人类免疫缺陷病毒(HIV,包括HIV-1和HIV-2)引起的获得性免疫缺陷综合征,已累计造成全球近3 300万人死亡,截止2019年底,全球仍有3 800万HIV感染者,AIDS的防治依然是一项严峻的全球性公共卫生问题[1]。目前上市的抗AIDS药物根据作用靶标可以分为反转录酶抑制剂、蛋白酶抑制剂、整合酶抑制剂、融合抑制剂及CCR5抑制剂[2]。随着高效抗反转录病毒治疗(highly active antiretroviral therapy,HAART)的普遍实施,AIDS的发病率和死亡率有所降低,然而由于RNA病毒聚合酶缺乏校正功能,导致病毒基因组突变率高,使临床药物作用降低或失效,因此研制新型的具有不同作用机制的抗AIDS药物一直是抗病毒药物研究领域的热点方向[3]。
由ViiV Healthcare开发的首创药物(first-in class)fostemsavir(Rukobia;FTR;BMS- 663068;GSK3684934)是HIV-1附着抑制剂temsavir(TMR;BMS-626529)的 前 药(图1),于2020年7月2日被美国FDA批准与其他抗反转录病毒药物联合使用,适用于因治疗耐药、不耐受或不安全而对当前HAART方案治疗失败成人的多重耐药HIV-1感染,推荐剂量为600 mg每天2次口服[4]。fostemsavir通过与HIV-1包膜糖蛋白gp160复合体的gp120亚基结合,选择性抑制病毒与细胞CD4受体的相互作用,从而防止病毒感染宿主细胞。本文就fostemsavir的作用机制、药效学、药动学、临床研究以及安全性等方面进行综述。
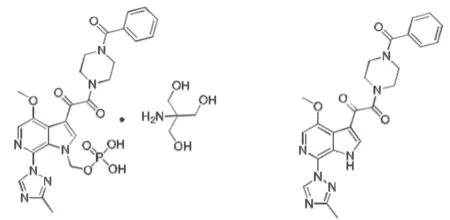
图1 fostemsavir与temsavir的化学结构式
1 作用机制
HIV-1包膜蛋白(Env)是由3个相同的糖蛋白单体构成的三聚体,每个单体含有受体结合蛋白gp120和跨膜融合蛋白gp41,以非共价相互作用连接。HIV-1感染细胞时,HIV-1 Env中的gp120首先必须与T细胞外表面上的膜受体CD4结合发生构象改变,随后与细胞表面的趋化因子受体CXCR4或CCR5结合,进而与细胞融合并侵入细胞内[5]。
fostemsavir是一种前药,口服后在肠道中被碱性磷酸酶转化为具有活性的temsavir,随后在小肠和升结肠中迅速吸收[6]。temsavir与HIV-1包膜上的gp120结合使其稳定在不被CD4受体识别的构象中,防止病毒与CD4+T细胞结合(图2)[7]。鉴于 HIV-1和 HIV-2之间包膜蛋白的结构差异,temsavir对HIV-2感染无抑制活性[8]。
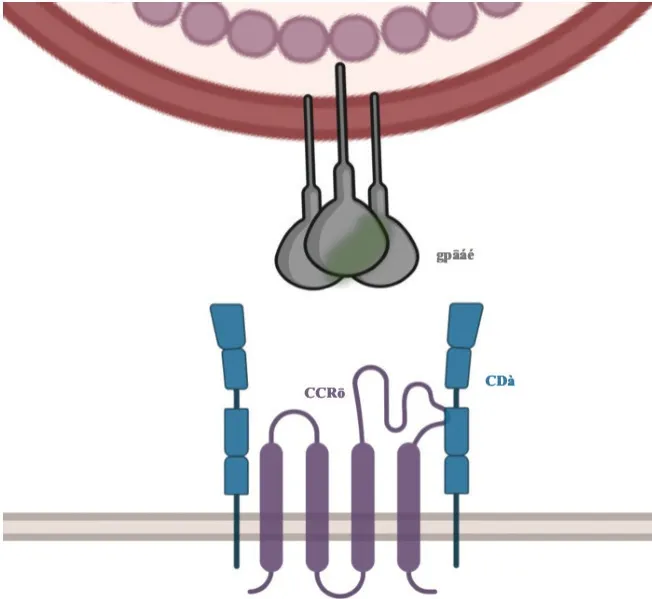
图2 temsavir的作用机制[9]
2 药效学
temsavir与gp120复合物的晶体结构显示(图3),其结合于gp120 内外域之间界面β20-21环下一个诱导型口袋,并与α1-螺旋的C末端相互作用,可稳定闭合的Env三聚体状态,抑制其构象改变,阻断与CD4结合,从而防止HIV-1附着,但不影响与其他抗体的结合[7-10]。

图3 temsavir(绿色)与 HIV-1 gp120结合的X-射线共晶结构(PDB code:5U7O)
temsavir在体外对BG505和BG505 T332N毒株的半数抑制浓度(IC50)为14 nmol/L,与Env三 聚 体BG505 SOSIP、DS-SOSIP的 结 合Kd值分别为73、87 nmol/L[7]。在针对多种实验室毒株和临床分离株的体外活性试验中,temsavir抑制CCR5嗜性毒株JRFL、SF-162和Bal的EC50值分别为0.4、0.5和1.7 nmol/L,抑制CXCR4嗜性毒株LAI、NL4-3、MN和IIIb的EC50值分别为0.7、2.2、14.8和16.2nmol/L,抑制双嗜性毒株PM1的EC50值是57.6 nmol/L,但对CXCR4嗜性毒株RF无活性(EC50>2 000 nmol/L)[11]。基因型分析temsavir对gp120包膜蛋白中4个位置的氨基酸 突 变(S375H/I/N/M/T、M426L/P、M434I/K和M475I)较敏感[12-13]。此外,交叉抗耐药性试验评估发现temsavir与其他HIV-1进入抑制剂无明显交叉耐药性,对巴利珠单抗、恩夫韦肽及马拉韦罗耐药的毒株均具有抑制活性,表明其可以与不同种类的进入抑制剂序贯或同时使用[14]。
3 药动学
单剂量和多剂量递增Ⅰ期临床研究发现血浆中检测不到fostemsavir,表明其快速转化为temsavir。temsavir最 大 浓 度(Cmax)和 浓 度-时间曲线下面积(AUC)值的增加均大于相应的给药剂量比例。temsavir的平均血浆半衰期为3.2~4.5 h(速释制剂)或7~14 h(缓释制剂),在2~3 d内达到稳态。temsavir与蛋白质高度结合(88.4%~92%),通过酯酶介导的水解和一小部分细胞色素P450(CYP)3A4介导的氧化途径进行代谢,主要代谢物通过尿液(44%~51%)、粪便(33%)和胆汁(5%)排泄[15]。temsavir的吸收率随高脂肪膳食增强,但在标准膳食中没有体现,并且安全性不会受到食物摄入的影响。因此,该药物可随餐服用或不随餐服用。表1提供了temsavir药动学的详细信息[16]。
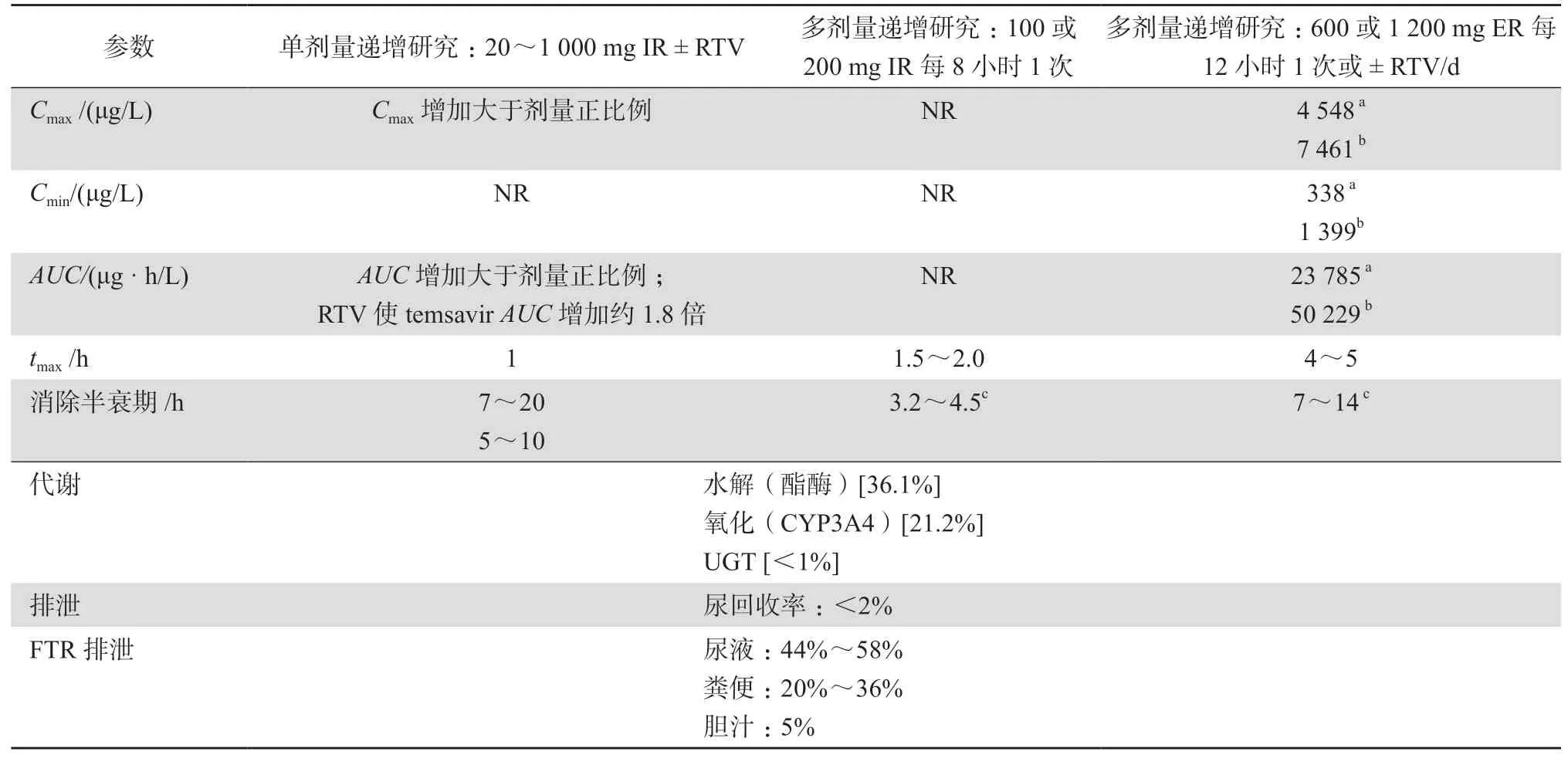
表1 不同给药方案的temsavir药代动力学数据
在一项针对HIV-1感染患者的Ⅱ期试验(NCT01009814)中,每12小时口服fostemsavir 600 mg联合利托那韦(RTV;ritonavir)100 mg(n= 9),在第8天时产生的temsavir平均Cmax和AUC0-24h值分别为2.24 mg/L和26.8 mg·h/L;每12小时1次口服fostemsavir 1 200 mg单药治疗,在第8天时产生的temsavir平均Cmax和AUC0-24h值分别为3.39 mg/L和42.6 mg·h/L[17]。
对于肾肝功能损伤的患者,单剂量给药fostemsavir 600 mg,对temsavir的药动学没有临床意义的影响。在肝肾功能正常的志愿者中,temsavir的蛋白结合率为81%,轻度、中度和重度肝功能不全的患者中temsavir的蛋白结合率分别为79.9%、81.9%和76.5%[18]。随着肝功能损害的严重程度增加,药物暴露参数值(Cmax和AUC0-inf)有增加的趋势,但对于轻度至重度肝功能不全的受试者,不需要调整fostemsavir的剂量。在具有轻度、中度或重度肾功能不全或患有终末期肾病(ESRD)且接受血液透析的HIV血清学反应阴性的患者中,口服fostemsavir 600 mg,temsavir的暴露量随着损伤严重性增加而增加,分类分析表明,严重肾功能不全患者的temsavir清除率降低了43%[14]。但对于轻度、中度或重度肾功能不全患者或ESRD患者,预计temsavir血浆浓度将保持在浓度-QTc分析定义的阈值以下,因此不需要调整fostemsavir剂量。temsavir不容易被血液透析清除,4 h血液透析大约清除12.3%的给药剂量,可以在不考虑血液透析时间的情况下将fostemsavir用于ESRD患者。总的来说,单次fostemsavir 600 mg在有肾或肝功能损害的受试者中是安全且耐受良好的,无需调整给药剂量[19]。
temsavir主要通过酯酶介导的水解途径代谢,经细胞色素P450(CYP)3A代谢的程度较低,是CYP3A、酯酶、P-糖蛋白(P-gp)和乳腺癌耐药蛋白(BCRP)的底物,诱导或抑制CYP3A、P-gp及BCRP的药物可能会影响temsavir血浆浓度。fostemsavir治疗剂量下与中度CYP3A诱导剂或强CYP3A、P-gp及BCRP抑制剂联合使用对temsavir血药浓度的影响不具有临床意义;与强CYP3A诱导剂联合给药会导致temsavir的浓度降低,例如强CYP3A4诱导剂利福平可使temsavir的Cmax和AUC分别降低76%和82%,可能导致病毒学应答丧失,因此禁止与强效CYP 3A诱导剂联用[20]。
此外,temsavir及其代谢产物是BCRP和有机阴离子转运多肽1B1/3(OATP1B1/3)的抑制剂,因此temsavir可能会影响作为OATP1B1/3或BCRP底物药物的药代动力学[21]。例如,fostemsavir可 增 加OATP1B1/3和BCRP底 物 瑞舒伐他汀的Cmax和AUC,与fostemsavir联用时需要减少剂量[20]。当fostemsavir与含有乙炔雌二醇/炔诺酮的口服避孕药合用时,可使乙炔雌二醇的暴露量增加40%,但并未显著影响炔诺酮水平,口服避孕药的乙炔雌二醇剂量不应超过每天30 μg[20]。temsavir对阿扎那韦、利托那韦、丁丙诺啡、纳洛酮、可比司他、达芦那韦、依曲韦林、法莫替丁、马拉韦罗、美沙酮、炔诺酮、拉替拉韦、利福布丁等药物药动学特征的影响没有临床意义[20]。
目前尚缺乏关于特殊人群(如老年人、孕妇或儿童)的数据,以及评估潜在药物相互作用的研究,尤其是与其他AIDS相关疾病治疗药物(如唑类)联用的研究[22]。
4 临床试验
fostemsavir上市前进行了3项关键的临床试验,确定了给药方案,并评估其在 HIV-1感染者中的有效性和安全性。表2总结了已发表的fostemsavir Ⅱ期和Ⅲ期临床试验[17,23-24]。
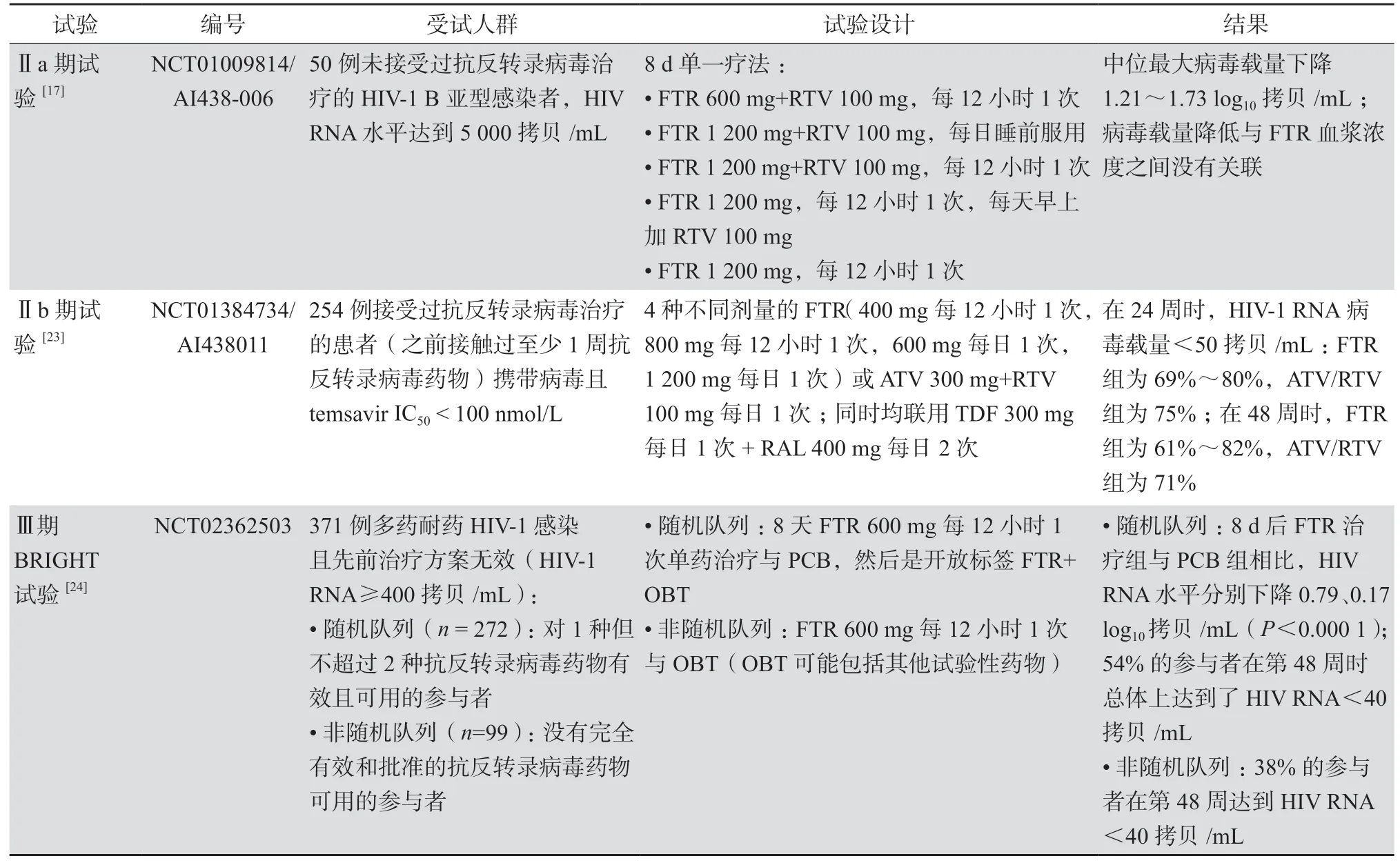
表2 fostemsavir的Ⅱ期和Ⅲ期临床试验
4.1 Ⅱ期试验
fostemsavir在两个随机Ⅱ期试验中进行了评估。第一项研究是在50例HIV-1 B亚型感染者中进行的开放标签、多剂量、平行Ⅱa期试验(NCT01 009814;AI438-006),参与者被随机分为5组(1∶1∶1∶1∶1),接受8 d不同剂量的fostemsavir(含或不含利托那韦),见表2:fostemsavir 600 mg+利 托 那 韦100 mg,每12小时1次;fostemsavir 1 200 mg+利托那韦100 mg,每日睡前服用;fostemsavir 1 200 mg+利托那韦100 mg,每12小 时1次;fostemsavir 1 200 mg,每12小时1次,每天早上加100 mg利托那韦;fostemsavir 1 200 mg,每12小时1次。在研究期间,不同方案组的患者血浆HIV-1 RNA载量中值均下降超过1.0 log10拷贝/mL。当基线IC50高于0.1 μmol/L的参与者被排除在分析之外时,观察到血浆HIV-1 RNA载量中值下降超过1.5 log10拷贝/mL[17]。
在具有治疗经验的HIV-1感染患者中进行的Ⅱb期剂量范围试验(NCT01 384734;AI438011)结果显示fostemsavir与阿扎那韦/利托那韦一样有效。患者被随机分配接受不同剂量:fostemsavir 400 mg每日2次(n= 50)、800 mg每日2次(n=
49)、600 mg每日1次(n= 51)、1 200 mg每日1次(n= 50)或阿扎那韦/利托那韦300 mg/100 mg每日1次(n= 51)治疗;此外所有患者还接受拉替拉韦400 mg每日2次和替诺福韦300 mg 每日1次的治疗[23,25]。在第24周,fostemsavir治疗组总病毒载量抑制率(<50拷贝/mL)为69%~80%,接受阿扎那韦/利托那韦治疗组为75%,差异无统计学意义。在接受fostemsavir治疗的受试者中,24周时的应答率与基线temsavir IC50无关。fostemsavir组的200例患者中有13例(7%)出现严重不良事件,阿扎那韦/利托那韦组的51例患者中有5例(10%)出现严重不良事件[16]。在第48周,所有实验组的总病毒载量抑制率(<50拷贝/mL)为61%~82%,而对照组为71%。初始病毒载量<100 000拷贝/mL受试者的治疗应答率较高[25]。
4.2 Ⅲ期试验
BRIGHT试验(NCT02362503)是一项部分随机、双盲、安慰剂对照的Ⅲ期研究,在23个国家的108个中心进行,参与者多为接受HIV治疗超过15年(71%)或在进入试验前曾接受过5种或更多种不同抗HIV治疗方案(85%)的患者。BRIGHT试验共入组了371例多重耐药HIV-1感染且先前治疗方案无效(HIV-1 RNA≥400拷贝/mL)的患者,被分配到随机队列组(n=272)及非随机队列组(n=99)两个队列[26]。见表2。
在随机队列组患者中,至少有1种FDA 批准的抗反转录病毒药物保持完全活性,这些患者以3∶1的比例进行随机分配,在其当前失败的治疗方案中加入fostemsavir(600 mg,每日2次)或安慰剂进行联合治疗8 d,然后服用fostemsavir加上优化的背景治疗(OBT)。与安慰剂相比,fostemsavir在8 d后显著降低了HIV-1 RNA水平(0.79对0.17 log10拷贝/mL,P<0.001)。在使用fostemsavir加OBT后,54%的参与者在48周时达到了低于40 拷贝/mL的HIV-1 RNA水平。在48周时,平均 CD4+T细胞增加为139/μL[26]。
针对目前已批准的抗反转录病毒药物治疗无效的患者被分配进入非随机队列组,并在第1天开始接受fostemsavir(600 mg,每日2次)加OBT。38%的受试者在48周时达到了低于40拷贝/mL的HIV-1 RNA水平,平均 CD4+T细胞增加为64/μL[26]。
第96周中期分析的终点包括血浆HIV-1 RNA低于40拷贝/mL的受试者比例,CD4+T细胞计数相对于基线的变化,不良反应的发生率及导致停药和死亡的不良事件。在随机队列组中,病毒学抑制率(HIV-1 RNA<40拷贝/mL)从第24周的53%(144/272)增加到第96周的60%(163/272)。非随机队列组中,第24周和第96周时应答率为37%(37/99)。重要的是,在研究进行的96周内,与基线相比两组的CD4+T细胞计数平均值均出现了显著且具有临床意义的增加:随机队列组中CD4+T细胞为205/μL,非随机队列组中为119/μL。最常见的不良反应(发生率≥5%,所有级别)是恶心和腹泻。很少有不良事件导致停药(26/371,7%),在第 96 周因不良事件而停止使用fostemsavir 治疗的参与者比例为 7%(随机组5%对非随机组12%)。随机队列组272例中有12例(4%)死亡,非随机分组的99例中有17例(17%)死亡;死亡者的平均基线CD4+T细胞计数为11/μL。结果表明,在经历过大量治疗、HIV-1疾病晚期且治疗选择有限的个体中,基于fostemsavir的抗反转录病毒疗法通常具有良好的耐受性,且在96周内病毒学和免疫学应答率均有所升高[24]。
5 安全性
fostemsavir的安全性较好,在Ⅱa期试验中,78%的治疗者(n=39)总体上至少发生了一次不良事件,在50 d的监测期内这一比例为 66%(33/50),没有受试者因为不良事件而停止治疗。最常见的不良事件包括头痛、皮疹和尿急,所有不良事件的强度都是轻度或中度(1级或2级),没有报告3级或4级不良事件[17]。Ⅱb期试验的96周安全性分析结果显示,在fostemsavir组和对照组中分别有2.5%和10%的治疗发生了导致停药的不良事件。fostemsavir组中共有91%的受试者至少发生1次不良事件(1~4级),而对照组为98%。最常见的不良事件包括感染(鼻咽炎、尿路感染、上呼吸道感染)、腹泻、恶心、呕吐和头痛[27]。
在Ⅲ期BRIGHTE试验中,fostemsavir组与安慰剂治疗组中归因于研究药物的不良事件发生率差异分别为19%和20%[24]。随机队列组接受fostemsavir加OBT的患者(n=271)中,发生率≥2%的不良反应(1~4级)包括恶心(10%)、腹泻(4%)、头痛(4%)、腹痛(3%)、消化不良(3%)、疲劳(3%)、皮疹(3%)、睡眠障碍(3%)、免疫重建性炎症综合症(2%)、嗜睡(2%)和呕吐(2%)。恶心发生率最高,但在fostemsavir组与安慰剂治疗组中的发生率相似(分别为6%和7%)。较不常见的不良事件(发生率为<2%的患者)包括QT间期延长(所有病例均为无症状)、肌痛、头晕、味觉障碍、周围神经病和瘙痒。报告的大多数不良事件(81%)为轻度或中度。由于不良事件而停止fostemsavir治疗的患者比例为7%(随机队列中为5%,非随机队列中为12%)。导致治疗终止的最常见不良事件大多与AIDS 相关事件或感染有关。3%的患者发生了严重的药物反应,其中包括3例严重的免疫重建性炎症综合征[20]。BRIGHTE试验随机队列组(n=271)中,发生率≥2%实验室异常(3~4级)最常见的是肝胆异常(24%)和肌酐升高(17%),包括:丙氨酸氨基转移酶水平>正常值上限(ULN)5倍(5%),天冬氨酸氨基转移酶水平>正常值上限5倍(4%),直接胆红素超过正常值上限(7%),总胆红素≥正常值上限2.6倍(3%),胆固醇≥300 mg/dL(5%),肌酐>正常值上限1.8倍或基线水平的1.5倍(19%),肌酸激酶≥正常值上限10倍(2%),血红蛋白水平<9.0 g/dL(6%),高血糖>250 mg/dL(4%),脂肪酶水平>正常值上限3倍(5%),低密度脂蛋白水平≥190 mg/dL(4%),中性粒细胞≤599/mm3(4%),三酰甘油>500 mg/dL(5%)和尿酸水平
>12 mg/dL(3%)[20]。
QT研究和暴露-反应模型显示,以推荐剂量fostemsavir 600 mg每日2次给药,不太可能导致QTc延长,即使同时使用可能会增加fostemsavir暴露的药物如抑制CYP3A4、BCRP和P-gp的药物及用于提高其他抗反转录病毒药物水平的药物增强剂等。然而,每日2次fostemsavir 2 400 mg的超治疗剂量可能会导致QTc延长[28]。在HCV或HBV合并HIV感染的患者中使用fostemsavir与肝脏转氨酶升高有关,因此建议在该类患者中监测肝功能[20]。关于妊娠期间使用fostemsavir的人体数据不足以充分评估与药物相关的出生缺陷和流产风险,在怀孕大鼠和兔子中的研究表明,在低剂量下没有发育毒性,但在较高剂量下观察到胎儿畸形[16]。
6 总结
fostemsavir是HIV-1附 着 抑 制 剂temsavir的前药,与其他抗反转录病毒药物联合用于治疗曾尝试过多种抗HIV疗法,并且由于耐药、不耐受或安全性的考虑而对当前抗反转录病毒药物方案治疗失败的多重耐药HIV-1成人感染者,推荐剂量为600 mg每天2次口服。Ⅲ期研究结果显示,大多数(60%)多重耐药HIV-1成人感染者接受fostemsavir和OBT后,可以维持病毒抑制直至96周,并且有较好的耐受性。总之,fostemsavir作为FDA批准的第一个针对HIV病毒周期中附着过程的药物,对其他种类的抗反转录病毒药物没有显现出明显的耐药性,将为多重耐药HIV-1感染的患者提供一种全新的治疗方法。
