黄精多指标成分近红外光谱快速定量分析模型建立
2022-12-05吴杭莎韦飞扬葛卫红杜伟锋李昌煜张叶峰
吕 悦, 吴杭莎, 韦飞扬, 葛卫红*, 杜伟锋,2,3*, 李昌煜, 张叶峰
(1.浙江中医药大学药学院,浙江 杭州 310053;2.浙江中医药大学中药炮制技术研究中心,浙江 杭州 311401;3.浙江中医药大学中药饮片有限公司,浙江 杭州 311400;4.浙江中医药大学中医药科学院,浙江 杭州 310053;5.宁波市中药饮片有限公司,浙江 宁波 315000)
黄精是我国应用历史悠久的传统名贵滋补中药[1]。黄精含有多糖、皂苷、黄酮、生物碱、木脂素[2-4]类化合物,其中多糖、皂苷类成分含量较高,为发挥药效作用的主要成分,有抗疲劳、抗氧化、降血糖血脂等作用[5-6],可作为体现黄精饮片质量的指标。5-羟甲基糠醛[7]为黄精炮制后产生的化合物且含量变化巨大,其使用安全风险和治疗保健功效存在争议。因不同产地的黄精配方颗粒中5-羟甲基糠醛含量差异较大也将其列为主要成分[8]。2020年版《中国药典》对黄精含量测定项下的指标为多糖,而黄精炮制前后多糖、皂苷、5-羟甲基糠醛含量都发生变化[9-11]。目前,黄精饮片常用色谱法开展质量评价,包括薄层色谱、高效液相色谱法、指纹图谱和液质联用法等手段[12-13]。基于绿色化学可持续发展理念,创新中药提取分析方法,构建绿色中药质量评价体系,建立一种快速无损,简便可靠的黄精饮片多指标定量方法尤为重要。
近红外光谱分析技术是通过照射红外光得到样品吸收光谱,反应样品中分子振动能级跃迁的信息,目前已广泛应用于食品、中药材真伪鉴别、产地鉴别及质量定量分析方面[14-15],是一种有效的质量控制和鉴别技术,有检测快速、操作简便、高灵敏度等特点,且在多种药材有所应用,如延胡索、片姜黄、薏苡仁[16-18]等。在此基础上,本研究首次利用近红外光谱技术进行黄精饮片的质量控制,通过收集大量不同产地黄精饮片,以多糖、皂苷和5-羟甲基糠醛含量为黄精活性成分代表,采集所有样品的近红外光谱图,结合偏最小二乘法(PLS)建立黄精饮片多指标近红外定量模型,以期实现黄精饮片质量快速评价及控制。
1 材料
Antaris Ⅱ型傅立叶变换近红外光谱仪、TQ Analyst 8.3.125分析软件、U3000高效液相色谱仪(美国ThermoFisher公司);UV-2450紫外-可见分光光度计(日本岛津公司);KQ-500DB型数控超声波清洗机(昆山市超声仪器有限公司);NT-xs105电子分析天平(0.01 mg,瑞士 Mettler Toledo公司);DFD-700电热恒温水浴锅(天津市泰斯特仪器有限公司);MIL-SYN型超纯水仪(美国Milipore公司)。
无水葡萄糖标准品(纯度99%,批号G6172-500)、人参皂苷Rb1(纯度99%,批号110704-202129)均购自中国食品药品检定研究院;5-羟甲基糠醛标准品(纯度>98%,批号B21832)购自上海源叶生物有限公司;乙醇、二氯甲烷均为色谱纯,甲醇为分析纯,水为超纯水。
138批黄精分别购自浙江中医药大学饮片有限公司、安徽亳州千草国药、山东博康药业、河北春开制药等16家国内中药饮片有限公司。
2 方法与结果
2.1 多糖含量测定 按2020年版《中国药典》中黄精含量测定项下的方法测定。以多糖质量浓度为横坐标(X),吸光度为纵坐标(A),得到回归方程A=20.682X+0.011 9(R2=0.998 9),在0.003 1~0.023 2 mg/mL范围内线性关系良好,精密度、重复性、稳定性试验RSD分别为0.43%、1.07%、0.36%;平均加样回收率100.87%。
2.2 皂苷含量测定
2.2.1 供试品溶液的制备 精密称取黄精粉末0.5 g置于具塞锥形瓶中,加入80%乙醇25 mL,每个样品平行3份。60 ℃超声60 min,重复提取2次,过滤,合并滤液至50 mL量瓶中,用80%乙醇定容。
2.2.2 标准曲线 精密称取人参皂苷Rb1对照品适量,甲醇溶解,得1.0 mg/mL对照品溶液。分别吸取对照品溶液0.06、0.12、0.18、0.24、0.30、0.36、0.42 mL于试管中挥干,加入0.2 mL 5%香草醛-冰醋酸溶液和0.8 mL高氯酸,摇匀,在60 ℃水浴保温15 min后,冰浴2 min,加入5 mL冰醋酸溶液,摇匀,静置5 min[19]。以相应试剂为空白,于568 nm波长测定吸光度。以人参皂苷Rb1质量浓度为横坐标(X),吸光度为纵坐标(A),得到回归方程A=13.704X-0.015 9(R2=0.995 2),在0.009 9~0.070 3 mg/mL范围内线性关系良好。
2.2.3 方法学考察 精密吸取对照品溶液,按“2.2.2”项下方法连续测定6次,RSD为0.78%。取同一批样品,按“2.2.1”项方法制备供试品溶液,按“2.2.2”项下方法分别于0、5、10、20、40、60 min测定,RSD为1.35%,表明供试品溶液稳定性良好。取同一批样品,按“2.2.1”项方法平行制备6份供试品溶液,按“2.2.2”项下方法测定,RSD为0.61%,表明方法重复性良好。取已知皂苷含量的黄精样品粉末6份,每份0.5 g,精密加入人参皂苷Rb1对照品,测定平均加样回收率为99.83%,RSD为1.12%。
2.2.4 含量测定 精密吸取供试品溶液0.1 mL,按“2.2.2”项下的方法测定,计算皂苷含量。
2.3 5-羟甲基糠醛含量测定
2.3.1 供试品溶液制备 精密称取样品1 g于具塞锥形瓶中,精密加入20 mL二氯甲烷,超声60 min,放冷,补重。精密吸取10 mL,蒸干,残渣加入甲醇溶解,转移置5 mL量瓶中,甲醇定容至刻度,过0.22 μm滤膜即得。
2.3.2 对照品溶液制备 精密称取19.37 mg 5-羟甲基糠醛于10 mL量瓶中,加入甲醇定容至刻度,精密吸取上述溶液置于10 mL量瓶中,加甲醇定容置刻度,摇匀,即得0.193 7 mg/mL对照品溶液。
2.3.3 色谱条件 Agilent ZORBAX-Extend-C18色谱柱(250 mm×4.6 mm,5 μm);流动相甲醇(A)-水(B),梯度洗脱(0~16 min,7%A;16~17 min,7%~20%A;17~40 min,20% A);检测波长300 nm;柱温30 ℃;进样量10 μL;体积流量1 mL/min[20]。色谱图见图1,各峰达到基线分离,分离度均大于1.5。
2.3.4 线性关系考察 精密吸取对照品溶液1、2、5、10、15、30 μL测定,以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y),得回归方程Y=45 317X+1.179 7(R2=0.999 3),在0.000 1~0.005 8 μg范围线性关系良好。
2.3.5 精密度试验 精密吸取对照品溶液,在“2.3.3”项下方法连续进样6次,测定峰面积,RSD为0.85%,表明仪器精密度良好。
2.3.6 稳定性试验 取同一批样品,在“2.3.1”项方法制备供试品溶液,在“2.3.3”项下方法分别于0、1、2、4、8、12、24 h进样10 μL,测定峰面积,RSD为0.72%,表明供试品溶液稳定性良好。
2.3.7 重复性试验 取同一批样品,在“2.3.1”项方法平行制备6份,按“2.3.3”项下方法进样分析,测定峰面积,RSD为1.68%,表明方法重复性良好。
2.3.8 加样回收率试验 取已知含量黄精粉末6份,每份1 g,精密加入5-羟甲基糠醛对照品在“2.3.1”项方法制备供试品溶液,测定平均加样回收率为99.12%,RSD%为0.74%。
2.4 近红外光谱采集 将138批黄精粉碎后过5号筛,取约10 g于石英杯中摊平,以内置背景做参比扣除,采集光谱图。采样方式积分漫反射,波数区间3 999~10 001 cm-1,分辨率8.0 cm-1。室温23~27 ℃,重复扫描3次,取其平均光谱,见图2。
2.5 校正集与验证集的划分 从138批黄精中随机选取16批作为验证集,122批作为校正集,含量测定结果见表1。
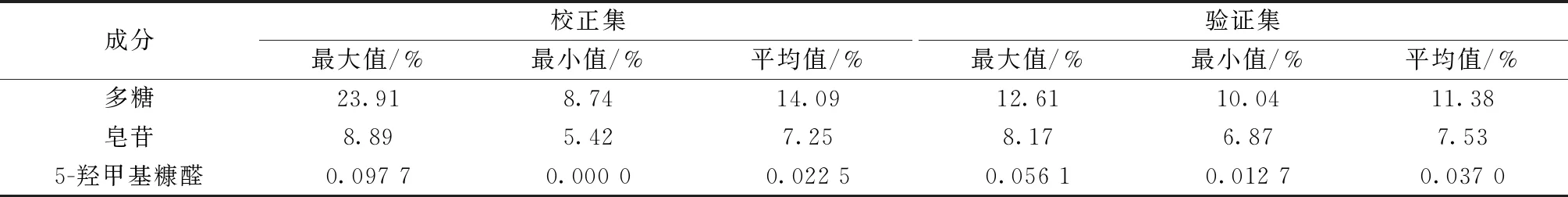
表1 校正集与验证集的含量测定结果
2.6 近红外光谱预处理 定量模型的校正算法有逐步多远线性回归法(SMLR)、偏最小二乘法(PLS)、主成分回归法(PCR)。对于组分复杂的中药,多选用PLS[21-22]。以相关系数、均方差、预测均方差为考察指标进行考察,其中相关系数越接近1,均方差、预测均方差越接近0,模型预测精密度越好。首先通过多元信号校正(MSC)或标准正则变换(SNV)对由于样品颗粒和分布不规则引起的偏差进行消除,减少光散射或光程差造成的影响[23];再通过原始光谱、一阶求导(1std)或二阶求导(2std)减轻光谱基线漂移;最后用无平滑(NS)、卷积平滑滤波(S-G)或者Norris导数平滑滤波(Nd)处理方法提高信噪比、减小随机误差[24],见表2。由表2可知,当多糖、皂苷、5-羟甲基糠醛分别选择MSC+1stD+NS、MSC+Spectrum+S-G、MSC+1stD+S-G方法处理时,相关参数最优,因此选用此方法对光谱数据预处理。

表2 不同光谱预处理方法对校正模型的影响
2.7 波段选择 以性能指数为指标,通过TQ Analyst软件优化,选择最适宜分析的光谱波段,见表3,得到多糖、皂苷、5-羟甲基糠醛最优波段分别为5 149.01~4 720.89、5 850.97~5 800.83、4 898.31~4 184.77、9 931.6~8 280.84 cm-1,模型预测精密度良好。

表3 不同光谱范围的影响
2.8 主因子数选择 通过PLS建立定量模型时,主因子数的数量会影响定量模型的精密度。主因子数过少可能会导致精密度降低,主因子数过多可能会产生过拟合现象。通过预测残差平方和以及内部交叉验证均方根误差(RMSECV)筛选主因子数,考察主因子数对多糖、皂苷、5-羟甲基糠醛的影响,结果表明最佳主因子数分别为6、9、5时模型的RMSECV最小,模型的准确度最高。
2.9 异常值剔除 经光谱Outliers优化,以马氏距离作为评价指标,对异常值进行剔除,以保证模型的准确性,见图3。异常值检验表明样品均符合要求。
2.10 定量模型的建立与验证 以筛选后的最优预处理方法作为参数评价指标(表4),建立定量模型,具体模型参数及各个变量实际值与预测值的相关性见图4~6。

表4 定量模型参数评价指标
将预测集(n=16)样品光谱输入定量模型中,得出其预测值,通过预测值与实际值的相对偏差,考察模型的准确度,见表5与图7。多糖、皂苷、5-羟甲基糠醛含量预测值与实际值的平均相对偏差分别为1.60%、0.87%、0.14%,均小于2%,t检验P值分别为0.92、0.84、0.86,均大于0.05,表明验证集预测值与检验值无显著性差异,模型准确度良好。

表5 验证集含量预测与结果偏差
2.11 定量模型方法学考察 精密度试验,在“2.4”项下方法采集近红外光谱图6次,导入建立的近红外定量模型中,预测多糖、皂苷、5-羟甲基糠醛含量,RSD均小于2%,表明该定量模型精密度良好。重复性试验,取同一批样品,分成6份,在“2.4”项下方法采集近红外光谱图,导入建立的近红外定量模型中,预测多糖、皂苷、5-羟甲基糠醛含量,RSD均小于2%,表明该定量模型重复性良好。稳定性试验,取同一批样品,分别于0、1、2、4、8、12 d,在“2.4”项下方法采集近红外光谱图,导入建立的近红外定量模型中,预测多糖、皂苷、5-羟甲基糠醛含量,RSD均小于2%,表明该定量模型稳定性良好。
3 讨论
本研究测定黄精的含量,通过近红外光谱法结合偏最小二乘法(PLS)建立黄精近红外多指标定量模型,建立模型的R值均大于0.9,相对偏差均小于2%,模型准确度良好,实现了近红外光谱技术在黄精饮片中的首次应用以及多糖、皂苷、5-羟甲基糠醛含量的快速测定,为其在炮制全过程质量控制奠定了基础。
