Fe(OH)x-Pt/BN催化剂上的巴豆醛加氢:Pt-Fe(OH)x相互作用促进不饱和醇选择性
2022-11-23叶艳文胡一鸣贾爱平李艳明鲁继青
叶艳文,胡一鸣,贾爱平,李艳明,王 瑜,鲁继青*
(1.浙江师范大学化学与生命科学学院,浙江 金华 321004;2.浙江师范大学图文信息中心,浙江 金华 321004)
α,β-不饱和醛选择性加氢制不饱和醇是生产医药、香料等各种精细化学品的重要工艺[1]。与NaBH4、LiAlH4等强还原剂相比,使用氢气(H2)作为加氢试剂是一种原子利用效率更高、更可持续的生产过程[2]。然而,该过程具有挑战性,因为副产物饱和醛比目标产物不饱和醇在热力学上更易生成。因此,需要高效的多相催化剂来实现不饱和醇的高选择性。

因此,Pt基界面位点的构建可能是获得不饱和醇高选择性较有前景的方法。从这个意义上说,这种界面位点的可控构建对于优化催化性能和构效关系的建立至关重要。与传统共沉淀法制备Pt基双金属催化剂[36-42]相比,湿化学方法可以更精确地控制界面位点的产生[43]。此外,众所周知,巴豆醛的化学吸附与催化剂的表面性质如表面酸碱性等因素密切相关,如果使用具有丰富表面酸碱性位点的氧化物,可能会干扰Pt基界面位点作用的研究。六方氮化硼(h-BN)含有非常有限的表面酸碱位点,而且其边缘可以有效地稳定活性物种的纳米颗粒[44],因此适宜用作活性物种的载体。
本文将氢氧化铁沉积在Pt纳米颗粒上以形成界面位点,并且在制备过程中通过Pt/Fe原子比来调节位点密度,制备的Fe(OH)x-Pt催化剂在巴豆醛加氢反应中具有更高的目标产物选择性。通过详细的催化剂表征和原位光谱解释了选择性提高的原因,为开发用于α,β-不饱和醛选择性加氢的高效催化剂提供了良好的借鉴作用。
1 实验部分
1.1 催化剂制备
FePt纳米颗粒制备:将20 mg的乙酰丙酮铂(Ⅱ)[Pt(acac)2,Pt含量大于质量分数49.6%,宇瑞(上海)化学有限公司)]溶解在9 mL油胺中[OA,C18含量为质量分数80%~90%,阿拉丁试剂(上海)有限公司],之后倒入50 mL的玻璃容器中,充入CO(99.999%)至100 kPa,并在180 ℃下加热40 min。然后,将溶解在2.4 mL OA中的5 mg乙酰丙酮铁(Ⅲ)[Fe(acac)3,质量分数大于99%,上海笛柏化学品技术有限公司]加入容器中,充入CO至100 kPa。将混合物加热至240 ℃并保持50 min,再冷却至室温。采用无水乙醇(质量分数大于99.7%,国药集团化学试剂有限公司)离心收集深色产物,并用无水乙醇和环己烷(质量分数大于99.7%,国药集团化学试剂有限公司)彻底洗涤和离心数次。最后将所得固体分散在环己烷中以备进一步使用。在制备过程中改变Fe(acac)3的量制备具有不同Fe/Pt物质的量比的纳米粒子。在不添加Fe(acac)3的情况下,按照与上述相同的步骤制备Pt纳米颗粒,所得样品表示为Pt/BN。在不添加Pt(acac)2的情况下按照与上述相同的程序制备不含Pt的Fe颗粒,所得样品表示为Fe/BN。
BN负载的FePt催化剂制备FePt/BN:将400 mg六方氮化硼[h-BN,质量分数大于99.0%,阿拉丁试剂(上海)有限公司]添加到20 mL含有FePt纳米粒子的环己烷中,Pt负载量约为质量分数2.5%。在真空干燥箱中处理悬浮液以除去溶剂。所得催化剂表示为xFePt/BN,其中x表示催化剂中的Fe/Pt物质的量比。如,0.1FePt/BN表示该催化剂中n(Fe)∶n(Pt)=0.1。
1.2 催化剂表征
催化剂中Pt和Fe的实际含量由美国赛默飞公司ARLADVANT’X Intelli Power 4200扫描X射线荧光(XRF)光谱仪测定。由于B和N等轻元素不能通过XRF技术可靠地测定,xFePt/BN催化剂中Pt和Fe的含量采用含有固定含量Pt和Fe的标准样品进行校准。
催化剂的X射线粉末衍射(XRD)分析在德国布鲁克公司Bruker D8 ADVANCE粉末X射线衍射仪上进行,Cu Kα,工作电压40 kV,工作电流40 mA,扫描范围2θ=10°~90°,扫描速率为0.15°·s-1。
高分辨率透射电子显微镜(HRTEM)和高角度环形暗场扫描透射电子显微镜(HAADF-STEM)为日本电子JEM F200,电压200 kV。催化剂中Pt的粒度分布通过统计每种催化剂中超过一百个Pt颗粒确定。
催化剂的CO化学吸附在日本麦奇克拜尔公司Belcat Ⅱ自动化学吸附仪上进行。将50 mg新制备的催化剂安装在反应器中,无需任何预处理,将CO脉冲注入催化剂并计算CO吸附量。
催化剂的氢气程序升温还原(H2-TPR)在自制装置上进行。将30 mg催化剂装入石英管反应器中,并在N2气流(99.99%,30 mL·min-1)中加热以去除表面污染物。样品冷却至30 ℃后,在5%H2-95%N2混合气(30 mL·min-1)中以10 ℃·min-1的升温速率将样品加热至700 ℃。升温过程中的信号由热导检测器(TCD)记录,实际H2消耗量通过已知量的CuO粉末还原进行校准。
采用美国赛默飞公司ESCALAB 250Xi型X射线光电子能谱仪(XPS)测定催化剂中各元素的化学状态,Al Kα(1 486.6 eV)。采用C 1s的结合能284.8 eV进行校正。
催化剂的CO化学吸附漫反射红外傅里叶变换(DRIFT)光谱在配备MCT检测器和PIKE DRIFT附件的美国赛默飞公司Nicolet iS50光谱仪上进行。催化剂装载在样品架中并暴露于10%CO-90%N2(20 mL·min-1)混合气中30 min。再用N2(30 mL·min-1)吹扫1 h后进行谱图采集,扫描次数为128次。
巴豆醛加氢的原位傅里叶变换红外(FT-IR)光谱研究在同一台Nicolet iS50光谱仪上进行。自支撑样品圆片(约30 mg,直径13 mm)装入石英池中,并用N2(30 mL·min-1)吹扫1 h。采用H2气流(99.999%,26 mL·min-1)通过保持在0 ℃的液体巴豆醛的饱和发生器引入气态巴豆醛。在N2吹扫(30 mL·min-1)30 min后,引入H2气流(26 mL·min-1)并将样品以5 ℃·min-1的升温速率加热到80 ℃,进行光谱采集(采用不同对应温度下的背景光谱)。
1.3 催化剂性能测试
巴豆醛(CRAL)的液相加氢在体积为50 mL的玻璃容器中进行。将催化剂(10 mg)装入容器中并与10 mL异丙醇(质量分数大于99.5%,国药集团化学试剂有限公司)和0.492 mmol CRAL混合。用H2气流冲洗容器3次以除去内部空气,之后充入160 kPa H2并加热至80 ℃开始反应。
CRAL的气相加氢反应在固定床反应器中进行。将100 mg催化剂装入内径8 mm的管状石英反应器中,在催化剂床层中间放置热电偶监测反应温度。采用H2气流(26 mL·min-1)通过含有0 ℃液体CRAL的饱和发生器,将CRAL蒸气引入反应器。进料中的CRAL分压为1.06 kPa,总反应压力为100 kPa。整个气体管线保持在50 ℃,以避免产物冷凝。反应物和产物的检测采用配备有火焰离子化检测器(FID)和DB-Wax毛细管柱的气相色谱仪(Shimadzu GC-2014)分析,碳平衡约为100±5%,主要产物有丁醛(BUAL)、巴豆醇(CROL)、丁醇(BUOL)和丙烯及部分聚合物。
2 结果与讨论
2.1 催化剂表征结果
表1总结了催化剂的物化性质。由表1可知,催化剂具有相似的表面积,约(15~16) m2·g-1,略低于BN载体的表面积18 m2·g-1。XRF测定催化剂中Pt的实际含量接近理论值(质量分数2.13%~2.72%),表明所有Pt纳米颗粒都成功沉积在BN表面上。然而,催化剂中Fe的实际含量低于制备理论值。如,Fe/Pt物质的量比为0.1的0.1FePt/BN催化剂是将理论上n(Fe)∶n(Pt)=0.3的Fe和Pt前体混合制备的,所得FePt固体中的Fe含量较少是由于CO对Fe前驱体的还原能力较弱,因此在洗涤过程中去除了大部分游离Fe离子。尽管如此,当添加更多的Fe(acac)3时,最终催化剂中Fe/Pt物质的量比增加,0.6FePt/BN上获得最高的Fe/Pt物质的量比0.64(理论值为1)。XPS表征得到的表面Fe/Pt比值远高于相应的体相含量比,这是因为Fe高度分散在Pt颗粒的表面上。然而,随着催化剂中Fe含量的增加,表面/体相比逐渐降低,表明Fe物种在高负载情况下发生了聚集的情况。催化剂的CO吸附量也总结在表1中,Pt/BN上的CO吸附量为1.31 mmol·gPt-1,对应的Pt分散度为0.25。由于Pt表面上Fe物种的修饰,CO吸附量随着Fe含量的增加而持续下降,说明暴露的表面Pt原子逐渐减少。如,0.1FePt/BN催化剂的CO吸附量为0.77 mmol·gPt-1,是Pt/BN(1.31 mmol·gPt-1)的59%,这意味着表面Pt原子的一半被Fe物种覆盖。对于0.6FePt/BN催化剂,它的CO吸附量非常低(0.014 mmol·gPt-1),表明催化剂中大部分表面Pt原子被Fe物种覆盖。因此,由于Fe物种的修饰作用,基于CO吸附量计算的含Fe的Pt催化剂中的Pt分散度数值可能具有较大的误差。尽管如此,CO吸附量很好地反映了这些催化剂中的Fe-Pt相互作用,并证实了Fe修饰的Pt纳米粒子的成功合成。

表1 PtFe/BN催化剂的物化性质
图1为xFePt/BN催化剂的XRD图。由图1可知,催化剂具有六方氮化硼(JCPDS No. 73-2095)的特征衍射峰以及较弱的金属Pt的衍射峰(JCPDS No. 87-0640)。
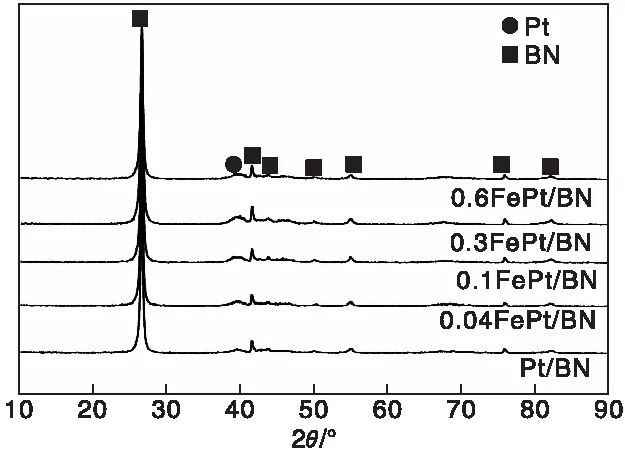
图1 xFePt/BN催化剂的XRD图Figure 1 XRD patterns of xFePt/BN catalysts
图2为Pt纳米粒子、Pt/BN、0.1FePt/BN、0.6FePt/BN催化剂的HRTEM照片及0.1FePt/BN催化剂的HAADF-STEM-EDX元素映射图像。图2及图S1(支持信息,详见电子版)清楚地表明,未负载的Pt纳米颗粒非常均匀,平均直径为4.1 nm。Pt纳米颗粒在BN上的沉积几乎不会改变Pt尺寸(4.3 nm),因为在Pt沉积后没有进行后处理。0.1FePt/BN和0.6FePt/BN催化剂中,Pt物种的平均直径(4.7~4.8) nm,略大于Pt/BN,可能是由于Fe物种对Pt纳米粒子的表面修饰。所有催化剂均显示出明显的Pt(111)平面(0.23 nm)条纹,但未观察到含铁物质。HAADF-STEM-EDX元素分析证实了催化剂中Fe物质的存在。以0.1FePt/BN催化剂为例,可以清楚地观察到Pt和Fe的均匀信号,并且Fe元素的密度低于Pt元素,与XRF表征的结果一致。

图2 Pt纳米粒子、Pt/BN、0.1FePt/BN、0.6FePt/BN催化剂的HRTEM照片及0.1FePt/BN催化剂的HAADF-STEM-EDX元素映射图像Figure 2 HRTEM images of Pt nanoparticles,Pt/BN,0.1FePt/BN,0.6FePt/BN catalysts and HAADF-STEM-EDX elemental mapping images of 0.1FePt/BN catalyst
采用XPS表征分析催化剂中Pt和Fe物种的化学状态,结果如图3所示。由图3和表1可知,所有催化剂都含有大部分金属Pt0物种(Pt 4f7/2结合能为71.2 eV),以及小部分Pt2+(Pt 4f7/2结合能为72.4 eV)和Pt4+(Pt 4f7/2结合能为74.9 eV)[45]。这些结果表明大部分Pt前体在制备过程中被还原。对于含铁的催化剂,Pt物种的结合能向高结合能处偏移(0.1~0.2) eV,并且催化剂中的Pt0浓度相对较低(表1),这意味着Fe-Pt相互作用和从Pt到Fe物种的电荷转移。对于Fe物种,结合能为710.2 eV和711.9 eV的峰表明存在Fe2+和Fe3+物种[43],表明部分Fe3+前体被还原为Fe2+。0.1FePt纳米粒子的O 1s XPS谱图(图S2,支持信息,详见电子版)具有以532.0 eV为中心的对称峰,表明存在表面羟基[46]。此外,结合能为530.0 eV和533.2 eV的峰表明存在晶格氧和表面吸附氧物种,这可能是由Pt和PtOx上的氧物种引起的,表明Fe物种为氢氧化物形式[Fe(OH)x,x=2或3],与文献的结论一致[43]。

图3 xFePt/BN催化剂中Pt 4f和Fe 2p的XPS谱图Figure 3 Pt 4f and Fe 2p XPS spectra of xFePt/BN catalysts
图4是BN载体和xFePt/BN催化剂的H2-TPR曲线。由图4可知,BN载体在测试温度范围内没有还原峰,而ω(Fe)=0.6%的Fe/BN还原峰大约在480 ℃,归属于BN上FeOx或Fe(OH)x物质的还原。Pt/BN催化剂在100 ℃有一个弱还原峰(α),归属于Pt氧化物的还原。Fe修饰后,α峰随Fe含量逐渐增加,并在(320~350) ℃处出现了另一个还原峰(β)。由于催化剂中Pt氧化物的含量非常接近(表1),因此增加的α峰可归属于由于氢气溢流效应而导致的Pt颗粒附近的Fe(OH)x物种的还原。而β峰与远离Pt颗粒的Fe(OH)x的还原有关。
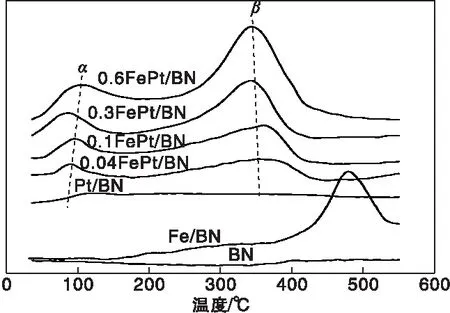
图4 BN载体和xFePt/BN催化剂的H2-TPR曲线Figure 4 H2-TPR profiles of BN support and xFePt/BN catalysts
图5是xFePt/BN催化剂上CO化学吸附的DRIFT谱图。由图5可知,Pt/BN催化剂在2 046 cm-1出现对称的强谱带,归属于金属Pt物种上的线性CO吸附[43]。此外,1 858 cm-1处的较弱的谱带可归属于CO在两个相邻金属Pt原子上的桥式吸附。Fe的加入导致红外谱图发生两个明显的变化。一方面,由于Pt表面原子被Fe(OH)x覆盖,峰面积随着催化剂中Fe含量的增加而逐渐下降,这与CO吸附量结果非常一致(表1)。Fe(OH)x的修饰也导致了1 858 cm-1处谱带的消失,表明Pt表面原子被Fe(OH)x物种隔离。另一方面,在(2 060~2 070) cm-1处的肩峰在Fe修饰的催化剂上清晰可见。由于Ptn+上吸附的CO通常在高于2 100 cm-1[47]的波数下观察到,因此该谱带不能归属于吸附在Ptn+物种上的CO,也不能归属于吸附在Fe(OH)x上的CO,因为Fe(OH)x/BN催化剂上没有CO吸附(图S3,支持信息,详见电子版)。因此,我们将2 070 cm-1处的谱带归属于吸附在Fe(OH)x-Pt界面处的Pt原子上的CO(如插图所示),界面Pt物种与金属Pt0相比电子云密度较低(处于缺电子状态)。此外,2 070 cm-1和2 046 cm-1谱带的面积比(表示为A2070/A2046)可用作界面Pt位点相对浓度的指标。如图5所示,0.1FePt/BN的重叠峰可以拟合为两个分量(请注意,0.1FePt/BN和Pt/BN中2 046 cm-1波段的半峰宽相同,即约为47 cm-1),A2070/A2046为0.126,表明与体相/表面Pt原子相比,界面Pt原子的比例非常有限(约13%)。

图5 xFePt/BN催化剂上CO化学吸附的DRIFT谱图Figure 5 DRIFT spectra of CO chemisorption on xFePt/BN catalysts
2.2 巴豆醛液相和气相选择性加氢反应性能

表2 xFePt/BN催化剂上液相CRAL加氢
在CRAL的液相加氢反应中,不同反应时间条件下,0.04FePt/BN催化剂和0.1FePt/BN催化剂上CRAL转化率、产物选择性及浓度如图6所示。
由图6可以看出,对于0.04FePt/BN催化剂,CRAL转化率在60 min内迅速增加并达到100%。同时,CROL和BUAL的选择性在60 min内持续下降(CROL从30%下降到20%,BUAL从55%下降到15%),而饱和醇BUOL的选择性上升(从小于20%上升到70%)。对产物浓度的分析清楚地表明,CROL和BUAL在反应40 min时达到最大值,随后下降,而BUOL的浓度不断增加。这种变化反映了CROL和BUAL进一步加氢形成BUOL的速度相当快。对于0.1FePt/BN催化剂,CRAL转化率在150 min达到100%。产物的选择性分为两个阶段,在100 min内(第1阶段),CROL选择性基本不变(70%~72%),BUOL选择性增加,BUAL选择性下降。以BUAL选择性的下降促使BUOL选择性的提高,这意味着BUOL的形成是由于BUAL的氢化造成的,而不是CROL的氢化。当反应在100 min后(第2阶段),对BUOL的选择性增加是由于CROL的消耗,从图中可以看到大约在100 min后,CROL的选择性开始下降,在160 min时选择性约43%。基于实验数据,0.1FePt/BN催化剂上CROL生成速率表达式可以推导为-dc/dt=kAcn,可转化为:
[1/(n-1)](1/cn-1-1/c0n-1)=kAt
式中,c是CRAL在一定反应时间的浓度(mol·L-1),c0是CRAL在反应混合物中的初始浓度(0.049 mol·L-1),kA是表观反应常数,n是CRAL的反应级数。由于H2分压在反应过程中保持恒定(160 kPa),因此不考虑氢气的影响。通过非线性回归,速率常数kA为0.34×10-3(mol·L-1)0.9·min-1,CRAL的反应级数为0.1。CRAL的低反应级数表明CRAL在催化剂上的高表面覆盖率,这是由于在有机介质(异丙醇)存在下CRAL容易吸附在催化剂表面上。
xFePt/BN催化剂上CRAL气相加氢反应结果见图7,反应温度80 ℃,空速15 600 mL·gcat-1·h-1。由图7可以看出,所有催化剂在反应初期失活,反应3 h后达到准稳态,失活是由于催化剂表面的积炭造成的[48]。在稳态下,Pt/BN催化剂的CRAL转化率约为42%,且CROL选择性仅为2%,主要副产物是BUAL(选择性约为90%)、BUOL(选择性约为5%)和丙烯(选择性约为3%)。相比之下,0.1FePt/BN催化剂的CRAL转化率为64%,CROL选择性高达82%。然而,催化剂中较高的Fe含量(如,0.6FePt/BN)导致CRAL转化率下降(约为6%),尽管CROL选择性仍然高于80%。催化剂在气相中的反应行为与在液相反应中相似,即Pt/BN催化剂具有活性,但对CROL没有选择性,而在催化剂中适量添加Fe(OH)x有利于提高活性和选择性。值得注意的是,0.1FePt/BN在气相中的性能与文献中的最佳结果相当(表S2,支持信息,详见电子版)。计算Pt/BN和0.1FePt/BN催化剂的活化能(Ea)以区分它们的本征反应性能(具体反应数据见表S3,支持信息,详见电子版),0.1FePt/BN催化剂的Ea约为43.0 kJ·mol-1,低于Pt/BN催化剂(约49.3 kJ·mol-1),证明0.1FePt/BN比Pt/BN具有更高的本征活性,与整体催化性能吻合。

图7 xFePt/BN催化剂上CRAL气相加氢反应Figure 7 Gas phase hydrogenation of CRAL overxFePt/BN catalysts
2.3 气相加氢的原位FT-IR研究

图8 Pt/BN和0.1FePt/BN催化剂上CRAL加氢的原位FT-IR谱图Figure 8 In situ FT-IR spectra of CRAL hydrogenation over Pt/BN and 0.1FePt/BN catalysts
2.4 讨 论
在过去的文献中,双金属催化剂(如Pt基和Ir基)是通过简单的共浸渍[14]或顺序浸渍[26-28,50-51]合成的。虽然可以实现双金属之间的相互作用,但单金属物种也存在于载体上,导致催化剂上活性物种较为复杂,对催化剂构效关系的建立带来了困难。本研究采用湿化学方法成功合成了一系列Fe(OH)x修饰的Pt纳米粒子。Fe(OH)x物种可以选择性沉积在尺寸均匀的Pt纳米颗粒表面上,并且这种修饰可以在制备过程中通过Fe/Pt比例进行精确控制。因此,可以确定Pt-Fe(OH)x相互作用并验证它们在反应中的作用。Fe(OH)x在Pt表面上的沉积导致Pt物种的变化,其对Pt表面的覆盖明显降低了暴露的Pt原子数目。另一方面,与Pt/BN催化剂相比,Pt-Fe(OH)x相互作用导致催化剂中形成更多的缺电子Pt物种(表1和图3),这种相互作用对催化剂微环境产生了重要影响,靠近Pt的阳离子Fe物种更容易通过氢的溢出而被还原(图4)。同时,CO化学吸附的DRIFTS结果(图5)清楚地揭示了Pt-Fe(OH)x周围的Pt物质电荷密度的变化。
现在,我们以定量方式区分不同Pt物种的活性。催化剂中的Pt物质包括远离Fe(OH)x和邻近Fe(OH)x的Pt原子,分别表示为Pt-Pt和Pt-Fe。基于表S3中的动力学结果、CO吸附(表1)和Pt-Pt和Pt-Fe位点的比率(图5),可以计算Pt物种的本征活性(转换频率,TOF)。对于Pt/BN,在70 ℃下,CROL生成速率为0.73×10-6mol·gPt-1·s-1,对应的TOF为0.56×10-3s-1。对于0.1FePt/BN,在70 ℃下,CROL生成速率为32.94×10-6mol·gPt-1·s-1,对应的TOF为42.78×10-3s-1。在0.1FePt/BN催化剂上获得的TOF包含来自Pt-Pt(TOFPt-Pt)和Pt-Fe(TOFPt-Fe)位点的贡献,因此TOFPt-Fe可以根据以下等式计算:42.78×10-3=TOFPt-Pt×[1/(1+0.126)]+TOFPt-Fe×[0.126/(1+0.126)],其中TOFPt-Pt为0.56×10-3s-1。基于图5中的IR结果,而因子1/(1+0.126)和0.126/(1+0.126)分别指Pt-Pt和Pt-Fe所占比例。因此,TOFPt-Fe为377.53×10-3s-1,这意味着Pt-Fe界面位点的本征活性比Pt-Pt位点高三个数量级(673倍),再次凸显了Fe(OH)x物种对反应性能的显著促进作用,这使得Fe(OH)x成为更有潜力的助剂。
3 结 论
Fe(OH)x修饰的Pt/BN催化剂对CRAL的加氢具有高活性和高选择性。Pt-Fe(OH)x相互作用导致界面Pt物种的电子性质发生变化,从而有利于羰基的活化并提高CROL的选择性。此外,需要适当的Fe(OH)x修饰以获得优化的性能,在本研究中,0.1FePt/BN催化剂上获得了最佳性能。而Pt表面上Fe(OH)x的过度修饰导致用于H2解离的Pt表面原子暴露较少,从而抑制了活性。这些发现揭示了Pt-Fe(OH)x界面在CRAL加氢反应中的关键作用,也可能为其他α,β-不饱和醛或共轭化合物的选择性加氢催化剂的设计提供一些借鉴。
