2‐乙基‐2‐芳基‐二氢喹啉衍生物的合成
2022-11-17朱超凡陈运荣
朱超凡,王 锐,陈运荣
(1.辽宁石油化工大学 石油化工学院,辽宁 抚顺 113001;2.上海科技大学 物质科学与技术学院,上海 201210)
喹啉及其衍生物广泛存在于一系列具有生理活性的天然产物与药物分子中[1‐2]。喹啉结构是重要的手性合成砌块,也是手性催化剂和配体的主要组成部分,因而喹啉衍生物的合成受到有机化学家和药物化学家的重视。治疗心律失常、抗菌与抗肿瘤且能够调节激素的Angustureine药物[3‐5]和抗疟药物——氨酚喹啉[6‐7]中包含喹啉结构;具有抗艾滋病病毒活性的药物——阿扎那韦[8‐9]中包含5,6,7,8‐四氢喹啉结构;从链霉素中提取的天然抗生素类药物virantmycins[10‐11]也以四氢喹啉结构为主要骨架。喹啉衍生物在有机不对称合成中也起重要应用,奎宁类催化剂是一类非常重要的有机小分子催化剂[12],同时一些奎宁衍生物还可以作为金属催化反应的手性配体[13]。
鉴于喹啉衍生物的重要应用价值,本课题组尝试一种新型喹啉衍生物的合成策略[14]。本文利用有机锂试剂对2‐苯基喹啉进行亲核加成,得到α烷基取代的二氢喹啉衍生物。此反应操作简便,为合成双取代二氢喹啉衍生物提供了新思路。
1 实验部分
1.1 试剂与仪器
试剂:二氯甲烷(DCM,体积分数为99.5%)、乙醚(Et2O,体积分数为95.0%)、乙酸乙酯(EA,体积分数为99.5%),上海科技股份有限公司;甲苯(PhMe,体积分数为99.8%)、无水硫酸钠(Na2SO4,质量分数为95.0%)、硅藻土(质量分数为95.0%),中国医药集团有限公司;2‐溴喹啉(C9H7Br,质量分数为99.5%),韶远化学科技有限公司;碳酸钾(K2CO3,质量分数为98.0%)、苯硼酸(C6H7O2B,质量分数为98.0%)、四氢呋喃(THF,体积分数为95.0%),上海毕得医药科技股份有限公司;四(三苯基膦)钯(PdP4C72H60,质量分数为98.0%)、2‐苯基喹啉(C15H11N,质量分数为98.0%)、乙基碘(EtI,体积分 数 为90.0%)、叔 丁 基 锂(t‐BuLi,体 积 分 数 为90.0%),上海安耐吉化学有限公司。除非另有说明,所有商业试剂无需进一步纯化即可使用。
仪器:Bruker Avance III HD光谱仪(FT,1H为400 MHz,13C为101 MHz),布鲁克(德国)科技有限公司;Agilent Technologies 6230 TOF LC/MS光谱仪,安捷伦科技有限公司;GreatWall DL SB‐10/20低温冷却液循环泵,郑州长城科工贸有限公司;bruker傅里叶变换红外光谱仪VERTEX 70V,上海尔迪仪器科技有限公司;JOANLAB实验室磁力搅拌器HS‐12数显加热恒温控温磁力搅拌机,群安实验仪器有限公司。
在氩气氛围下,通过活化的氧化铝柱纯化二氯甲烷、甲苯、醚、四氢呋喃,使用黄海硅胶HSGF254 TLC板对反应混合物进行薄层色谱(TLC)分析,并在紫外光下或通过钼酸铈铵或高锰酸钾染色进行可视化。快速柱色谱法在300~400目黄海硅胶HHGJ‐300上 进 行。使 用Bruker Avance III HD光谱仪记录核磁共振(NMR)光谱,1H和13C化学位移以 标 准 溶 剂 峰 为 基 准(CHCl3:δH=7.26,δC=77.16;CD3OD:δH=3.31,δC=49.00;(CD3)2CO:δH=2.05,δC=29.84)。使 用以下 字母报告多重性:s=单重态,d=双重态,t=三重态,q=四重态,m=多重态,br=宽共振。红外光谱数据由bruker傅里叶变换红外光谱仪VERTEX 70V测出,质谱数据从Agilent Technologies 6230 TOF LC/MS光谱仪以电喷雾电离(ESI+)模式获得。
1.2 实验方法
化合物1的合成:称取2‐溴喹啉(1 g)、碳酸钾(2 g)及苯硼酸(887 mg)于圆底烧瓶中,加入磁子,再加入四(三苯基膦)钯(285 mg),抽换氮气3次,在氮气保护下,将四(三苯基膦)钯(6.0 mL)、水(6.0 mL)及超干甲苯(35.0 mL)加入圆底烧瓶中,进行鼓泡处理15 min,在氮气保护下,将反应体系加热到80℃,当TLC板或LC‐MS(液相色谱‐质谱,用来检测相对分子质量)检测反应完成时,加水淬灭,用乙酸乙酯(30.0 mL)萃取3次,合并有机相,有机相用饱和食盐水(10.0 mL)洗涤,用无水硫酸钠干燥,硅藻土过滤,浓缩有机相。进行柱层析(V石油醚/V二氯甲烷=6∶1),得到化合物1(964 mg),其产率为97%。化合物1的合成路线见图1。

图1 化合物1的合成路线
化合物2(乙基锂)的合成:称取乙基碘(1.2 mL)于圆底烧瓶中,加入磁子,抽换氮气3次,在氮气保护下,加入超干四氢呋喃(28.0 mL),将反应体系冷却至-78℃。在此温度下搅拌30 min,缓慢加入叔丁基锂(28.0 mL),缓慢升至室温,在室温下搅拌2 h,得到乙基锂的四氢呋喃溶液(0.25 mol/L)。化合物2的合成路线见图2。
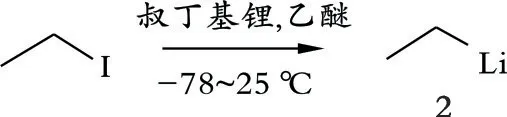
图2 化合物2的合成路线
化合物3的合成:以化合物1为底物,加入化合物2(乙基锂),在合适的溶剂与温度条件下发生亲核加成反应。化合物3的合成路线见图3。

图3 化合物3的合成路线
2 结果与讨论
2.1 实验条件的优化
2.1.1 亲核加成反应溶剂的筛选 在氮气氛围下,将2‐苯基喹啉(100 mg)溶于溶剂,在0℃下缓慢滴加乙基锂(6.0 mL),所用溶剂为超干溶剂。采用不同溶剂时亲核加成反应产物的产率见表1。

表1 采用不同溶剂时亲核加成反应产物的产率
由表1可知,分别以二氯甲烷和乙酸乙酯为溶剂时,反应无法进行;分别以甲苯和乙醚为溶剂时,产物的产率仅为10%和40%;以四氢呋喃为溶剂时,产物的产率最高,其值为70%。因此,最佳溶剂为四氢呋喃。
2.1.2 亲核加成反应温度的筛选 在氮气氛围下,将2‐苯基喹啉(100 mg)溶于四氢呋喃,在不同反应温度下缓慢滴加乙基锂(6.0 mL),不同反应温度下亲核加成反应产物的产率见表2。由表2可知,当反应温度为-78、-40、-20℃时,产物的产率分别为48%、55%、65%;当反应温度为0℃时,产物的产率为70%。因此,最佳反应温度为0℃。

表2 不同反应温度下亲核加成反应产物的产率
2.1.3 亲核加成反应的乙基锂体积筛选 在氮气氛围下,将2‐苯基喹啉(100 mg)溶于四氢呋喃中,在0℃下缓慢滴加乙基锂,乙基锂体积不同时亲核加成反应产物的产率见表3。

表3 乙基锂体积不同时亲核加成反应产物的产率
由表3可知,当乙基锂体积为2.0、3.0、4.0、5.0、6.0 mL时,产物的产率分别为20%、36%、57%、62%、70%。因此,乙基锂的最佳体积为6.0 mL。
综上,得到的最优条件为:2‐甲苯喹啉质量为100 mg,乙基锂体积为6.0 mL,反应温度为0℃,溶剂为四氢呋喃。
2.2 底物的拓展
在2‐甲苯喹啉质量为100 mg、乙基锂体积为6.0 mL、反应温度为0℃的条件下,以四氢呋喃为溶剂,进行底物的拓展实验。
称取化合物1(964 mg)置于圆底烧瓶中,加入磁子,抽换氮气3次;在氮气保护下,加入超干四氢呋喃(24.0 mL);将反应体系冷却至0℃,搅拌15 min,在氮气保护下,将乙基锂(57.0 mL)缓慢加入反应体系,然后将反应体系缓慢升至室温,在室温下搅拌2 h;当TLC板或LC‐MS检测反应完成时,将反应体系冷却至0℃,加入饱和的氯化铵水(10.0 mL)溶液淬灭;用乙酸乙酯(30.0 mL)萃取3次,合并有机相,有机相用饱和食盐水(10.0 mL)洗涤,无水硫酸钠干燥,硅藻土过滤,浓缩有机相。进行柱层析(V石油醚/V二氯甲烷=6∶1),得到产物3(774 mg),其产率为70%。该产物为白色油状物。化合物3的最佳合成路线如图4所示。
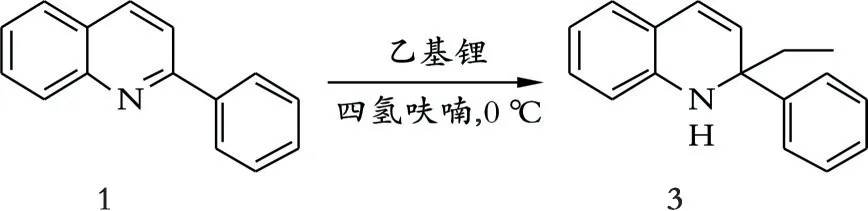
图4 化合物3的最佳合成路线
化合物4、化合物5的合成路线与化合物3的合成路线基本一致,化合物4及化合物5的合成路线如图5、6所示。化合物4为黄色油状物,其产率为75%;化合物5为黄色油状物,其产率为72%。

图5 化合物4的合成路线

图6 化合物5的合成路线
2.3 化合物的结构与表征
化合物3的核磁共振氢谱及碳谱如图7所示。由图7(a)可知,化合物3的核磁共振氢谱数据为:1H NMR(400 MHz,CDCl3):δ7.47~7.45(m,2H),7.36~7.32(m,2H),7.26~7.20(m,1H),7.00~6.98(m,1H),6.89~6.87(m,1H),6.59~6.55(m,1H),6.51~6.49(m,1H),6.40~6.38(m,1H),5.58~5.56(m,1H),4.03(s,1H),2.13~2.04(m,1H),1.95~1.86(m,1H),1.02~0.98(m,3H)。
由图7(b)可知,化合物3的核磁共振碳谱数据为:13C NMR(101 MHz,CDCl3):δ148.74,143.36,129.02,128.58,128.39,127.03,126.72,125.42,124.58,119.31,117.04,112.23,60.93,35.61,8.77。高分辨质谱数据:产物相对分子质量的实际值为236.143 1,理论值为236.143 4。分析结果表明,所得产物与目标产物一致。

图7 化合物3的氢谱及碳谱
化合物4的核磁共振氢谱及碳谱如图8所示。由图8(a)可知,化合物4的核磁共振氢谱数据为:1H NMR(500 MHz,CDCl3):δ7.39~7.37(m,2H),7.00~6.99(m,1H),6.98~6.97(m,3H),6.88~6.57(m,1H),6.56~6.54(m,1H),6.48~6.36(m,1H),5.53~5.51(m,1H),3.97(s,1H),3.79(s,3H),2.08~2.01(m,1H),1.92~1.85(m,1H),1.01~0.98(m,3H)。由图8(b)可知,化合物4的核磁共振碳谱数据为:13C NMR(126 MHz,CDCl3):δ158.30,143.34,141.13,128.96,128.58,126.98,126.63,124.26,119.23,116.92,113.83,112.14,60.44,55.38,35.47,8.80。高分辨质谱数据:产物相对分子质量的实际值为266.153 9;理论值为236.143 4。分析结果表明,所得产物与目标产物一致。

图8 化合物4的氢谱及碳谱
化合物5的核磁共振氢谱及碳谱如图9所示。由图9(a)可知,化合物5的核磁共振氢谱数据为:1H NMR(500 MHz,CDCl3):δ7.28~7.24(m,1H),7.04~7.01(m,3H),6.99~6.87(m,1H),6.76~6.74(m,1H),6.58~6.56(m,1H),6.55~6.51(m,1H),6.49~6.37(m,1H),5.58~5.56(m,1H),4.02(s,1H),3.78(s,3H),2.09~2.02(m,1H),1.91~1.84(m,1H),1.00~0.97(m,3H)。由图9(b)可知,化合物5的核磁共振碳谱数据为:13C NMR(126 MHz,CDCl3):δ159.80,150.47,143.30,129.59,129.02,128.28,127.03,124.66,119.33,117.74,117.09,112.27,112.02,111.36,60.90,55.32,35.65,8.76。高分辨质谱数据:产物相对分子质量的实际值为266.153 6,理论值为266.153 9。分析结果表明,所得产物与目标产物一致。

图9 化合物5的氢谱及碳谱
化合物3—5的红外谱如图10—12所示。由图10可以看出,3 392.57 cm-1处为N—H伸缩振动峰;742.62、698.12 cm-1处为单取代芳环弯曲振动峰;3 056.00 cm-1处为芳环中C—H的伸缩振动弱峰;2 966.44、2 930.38 cm-1处为烷基中C—H伸缩振动峰,以上证明有化合物3的特征峰。

图10 化合物3红外谱图
由11可以看出,3 387.67 cm-1处为N—H伸缩振动峰;825.78 cm-1处为对二取代芳环的弯曲振动峰;3 033.04 cm-1处为芳环中C—H的伸缩振动弱峰;2 964.00、2 931.69 cm-1处为烷基中C—H的伸缩振动峰;1 248.78 cm-1处为Ar—O的伸缩振动峰,以上证明有化合物4的特征峰。
由12可以看出,3 386.47 cm-1处为N—H的伸缩振动峰;775.35、696.53 cm-1处为间二取代芳环的弯曲振动峰;2 963.40、2 932.01 cm-1处为烷基中C—H的伸缩振动峰;1 248.15 cm-1处为Ar—O的的伸缩振动,以上证明有化合物5的特征峰。

图11 化合物4红外谱图
3 结 论
2‐乙基‐2‐芳基‐二氢喹啉衍生物的合成共分为三步。第一步,2‐溴喹啉与苯硼酸进行Suzuki交叉偶联;第二步,叔丁基锂与碘乙烷进行卤锂交换;第三步,合成的乙基锂亲核加成到喹啉的α位,将碳氮键断开,得到α,α‐双取代二氢喹啉产物,产率高达70%。此方法是目前首个直接将有机锂试剂亲核加成得到二氢喹啉的研究,其意义深远。

图12 化合物5的红外谱图
