含硫化合物极压作用的分子模拟
2022-11-16李义雅段庆华
李义雅, 赵 毅, 苏 朔, 龙 军, 段庆华
(中国石化 石油化工科学研究院,北京 100083)
高温(温度高于500 ℃)高载荷的苛刻工况下,润滑油处于边界润滑状态,润滑油膜不能防止摩擦副表面微凸体的接触,润滑油中的基础油和摩擦改进剂等组分难以发挥出保护摩擦副的作用,极压抗磨剂会在该工况下与摩擦副反应生成强度较大的表面膜,起到抗磨抗烧结作用。硫类化合物是被发现最早的润滑油极压添加剂,主要包括硫化烯烃、硫化脂肪酸酯、二硫化物、多硫化物等,最常用的是硫化异丁烯。含硫化合物具有优异的极压性能,含硫极压剂的研究对研究苛刻工况下油品的应用具有重要意义。
最早广泛应用的硫类添加剂是硫化猪油(SLO),是一种硫化动物三酸甘油酯。1939年,研究人员发现硫化鲸鱼油(SSWO)更易溶于石蜡基础油中,并比SLO有更好的热稳定性,这是润滑油历史上最重要的发现之一。SSWO热稳定性的提高源于其单酯结构,而SLO为三酯结构。借助现代表面分析方法,目前一般认为含硫表面膜的主要组成是FeS,同时还含有FeS2、Fe2O3、FeSO4、Fe2(SO4)3等,其硬度较大,剪切强度低,熔点高(FeS、FeS2分别为1193、1171 ℃),抗烧结能力好,抗磨性能差,因此含硫化合物是性能优异的极压剂。
对于含硫化合物与金属表面作用形成该表面膜过程,Allum等[1-3]和Forbes等[4-5]做了很多研究,并提出了含硫添加剂先吸附后反应的机理。主要过程为二硫化物先吸附在铁表面,在抗磨区S—S键断裂形成硫醇化铁,但是键的断裂是在吸附前还是吸附后尚未明确;当载荷达到极压条件时,接触区的温度会更高,因此C—S键断裂形成无机的含硫层。Plaza等[6-7]对二苯基二硫化物(DPDS)和二苄基二硫化物(DBDS)研究发现,二硫化物在边界润滑条件下形成表面膜的过程很复杂,首先发生S—S键的断裂,含苄基自由基中C—S键的活性较高,容易断裂形成元素硫,进一步反应生成硫化铁;而含苯基自由基中的C—S键较强,自由基活性较高,会与烃类溶剂中的氧等继续反应,且副产物会进一步引发各种次级反应,反应产物再与金属反应,生成苯磺酸盐、苯基硫碳酸盐等,发挥抗磨作用。但Najman等[8]采用X射线吸收近边结构谱(XANES)和原子力显微镜(AFM)研究含硫化合物的作用,认为二硫化物在摩擦作用下先分解出活性硫;在抗磨条件下,活性硫与铁或Fe2O3、FeO反应形成FeS2,并且可能与环境中的氧继续反应生成Fe2(SO4)3或FeSO4;在极压条件下,活性硫与铁反应生成FeS,且在抗磨条件生成的FeS2也可能会在更苛刻的条件下分解生成FeS。之前的研究多是根据反应产物组成推测反应机理,且实验研究及分析表征方法[8-10]难以验证详细的反应机理。
分子模拟是随着量子化学理论和计算机技术进步而发展的一种计算化学方法,可以研究物质在原子、分子水平的物理和化学性质,在物质的物理、化学作用机理领域已经有了一定应用[11-17]。基于计算机模拟方法,Wang等[18]利用密度泛函理论(DFT)研究了14种二烷基二硫化物的几何结构优化和电子结构,根据前线轨道理论和边界润滑理论,这14种二硫化物的活性原子是S原子和与之相连的C原子,活性键是S—S键和C—S键。依据吸附作用越强抗磨作用越强的原理,通过研究二硫化物与金属表面的化学吸附能,预测了14种含硫化合物的抗磨性能,且二苄基二硫化物的极压性能比二苯基二硫化物好。目前,对含硫化合物与金属表面作用形成表面膜作用机理尚没有统一结论,且对含硫表面膜能够在高温下发挥抗烧结性能的原因缺乏深入理解,需要进一步研究。
笔者采用量子化学方法,以二乙基二硫化物为模型分子,研究含硫化合物与Fe(110)表面的化学反应过程,考察含硫表面膜具有抗烧结性能的原因。
1 分子模拟方法
1.1 计算模型和参数
含硫极压剂中二硫化物的应用最广泛,为提高量子化学的计算效率,选择简单的模型分子来研究化学作用,用二乙基二硫化物为模型化合物,分子结构如图1所示。

图1 二乙基二硫化物的分子结构Fig.1 Molecular structure of diethyl disulfide
所有的分子模拟工作均采用BIOVIA公司的Materials Studio 8.0软件完成。金属表面选择Fe(110)表面,搭建周期性金属表面,为避免上下2个周期性结构的分子有相互作用,在构建的较小Fe(110)表面上添加2 nm的真空层,模型如图2所示。

图2 Fe(110)表面模型Fig.2 Model of Fe(110) surfaces(a) Vertical view; (b) Side view
使用DMol3模块的量子化学方法研究化学作用,为选择适用于研究体系的泛函方法,结合所研究的含金属表面周期性体系特征,尝试分别选择基于局域密度近似(LDA)的PWC泛函方法与基于广度梯度近似(GGA)的PBE泛函方法,对含二乙基二硫化物分子与Fe(110)表面的体系进行优化。分别采用这2种方法优化后,模型构象无明显异常,体系可收敛,2种泛函方法均可适用于所研究的含金属周期性体系,而GGA-PBE泛函方法的精确度更高,故其进行量子化学计算。对于计算过程所用精度,分别采用Coarse、Medium和Fine精度进行模拟计算。结果发现,采用Fine精度时,由于含有金属层表面,计算电子较多,所研究体系不收敛,平衡计算精度与可收敛性,选择Medium精度。
因此,最终确定量子化学采用基于广度梯度近似(GGA)的PBE泛函方法,k-point选择(2×2×1),在DND基组水平上进行全电子计算。能量、受力和位移的收敛标准分别为5.25×10-2kJ/mol、105.02 kJ/(mol·nm)和5×10-4nm,自洽场(SCF)迭代收敛的阈值设为2.63×10-2kJ/mol。
使用CASTEP模块的量子化学研究体系中原子间的电子变化,量子化学采用基于广度梯度近似(GGA)的PBE泛函方法,k-point选择(2×2×1),价单子(即最外层电子)与核的相互作用采用Ultrasoft赝势来描述, SCF迭代收敛的阈值设为2.63×10-2kJ/mol。
1.2 模拟过程
硫化物与Fe(110)表面的化学作用过程包括多个基元反应,且可能有不同的反应路径,因此先设计不同作用过程的基元反应,再进行模拟计算。对图2的金属表面模型,固定下面2层Fe原子,用DMol3优化含真空层的Fe(110)表面。用同样的参数对各基元反应的反应物和产物进行优化,将优化后的稳态构型分别作为各基元反应的初态和终态,采用完全线性同步和二次同步变换Complete LST/QST 方法搜索各基元反应的过渡态,用振动频率分析过渡态结构,再采用TS Optimization对过渡态进行优化,确定过渡态结构。
对优化后的结构,用CASTEP模块的Population analysis分析S原子与Fe原子的电子重合百分率,用键级的值表示。计算化学作用过程中S原子上的电荷量和反应中电荷的变化情况。用式(1)计算化学作用过程中的C—S键的键能(EBD(C—S)),其中E(C—S)为分子的能量,E(C·)和E(S·)为化学键均裂后2部分自由基的能量,单位为kJ/mol。
EBD (C—S)=E(C·)+E(S·)-E(C—S)
(1)
2 结果与讨论
2.1 硫化物与Fe(110)表面的反应机理
2.1.1 二乙基二硫化物与Fe(110)表面的反应机理
根据现有认识,二乙基二硫化物与Fe表面的作用机理包含2种基元反应历程,如图3所示。吸附反应历程中,二乙基二硫化物先与Fe(110)表面发生物理吸附,继而发生化学吸附,之后C—S键断裂,生成FeS。分解反应历程中,二乙基二硫化物中的S—S键先断裂,生成CH3CH2S·自由基,之后C—S键断裂,生成活性硫,硫与Fe(110)表面反应生成FeS。

R—CH3CH2·图3 二乙基二硫化物与Fe表面作用的2种反应历程Fig.3 Two reaction processes of diethyl disulfide with Fe surface
二乙基二硫化物与Fe(110)表面先吸附后反应作用过程中各个结构的能量大小和构象如图4所示。

C atom; H atom; Fe atom; S atom图4 二乙基二硫化物吸附/反应机理过程中体系的相对能量和构象Fig.4 Relative energy and conformation of the system in the diethyl disulfide adsorption process and reaction mechanism
根据图4中的能量和构象观察到,二乙基二硫化物在Fe(110)表面的作用下,S原子与Fe原子之间的距离逐渐接近,并且靠近Fe(110)表面时硫化物中S—S键断裂,最终CH3CH2S·吸附在Fe(110)表面,因此二乙基二硫化物先通过解离/吸附过程与Fe(110)表面发生作用,该过程的能垒为155.05 kJ/mol。之后,吸附在Fe(110)表面单硫化物中的C—S键断裂,C—S键断裂后生成FeS,2个C—S键断裂的能垒分别为209.00和178.62 kJ/mol,即为2个C—S键的键能。
二乙基二硫化物先分解生成活性硫,活性硫再与Fe(110)表面反应过程中各个结构的能量大小和构象如图5所示。由图5可以看到,二乙基二硫化物先发生S—S断裂生成单硫化物自由基,该过程的能垒是279.82 kJ/mol。单硫化物自由基经过发生C—S键断裂,生成活性S原子,能垒为306.30 kJ/mol。活性硫与Fe(110)表面反应生成FeS,该过程的能垒较低,只有66.93 kJ/mol,说明活性硫与Fe表面的反应很容易发生。

C atom; H atom; Fe atom; S atom图5 二乙基二硫化物分解反应机理过程中体系的相对能量和构象Fig.5 Relative energy and conformation of the system in the diethyl disulfide decomposition process and reaction mechanism
比较二乙基二硫化物与Fe表面吸附反应与分解反应2种作用机理的过程和能垒,如图6所示,红色数字代表基元反应的能垒。由图6可知,分解反应过程中生成的活性硫与Fe(110)表面反应的能垒最小,但是二乙基二硫化物分解生成活性硫的2步反应能垒都较大,说明二乙基二硫化物较难分解生成活性硫。如果生成活性硫,则活性硫很容易与Fe(110)反应,这也可以解释活性硫对金属表面的腐蚀作用。吸附反应过程中能垒最大的是解离/吸附过程,与二乙基二硫化物分解生成活性硫的能垒相比小很多,说明吸附反应过程较容易发生。所用DMol3分子模拟方法中,温度均在0 K条件下计算,且与实际温度无法直接对应,根据相同条件下的模拟计算,相对比较不同反应进行的难易。因此,根据2种反应历程中能垒的比较,在边界润滑状态下,工况较温和时,二硫化物更容易先与Fe表面发生化学吸附,S—S键在吸附时先断裂,进而C—S键断裂,生成FeS;工况较苛刻时,能够使二硫化物中的S—S、C—S键断裂,生成的活性硫很容易与Fe表面反应,生成FeS,即在高温下,以分解反应历程为主。
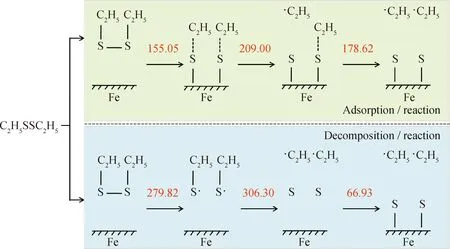
Red number—Energy barrier of the reaction, kJ/mol图6 二乙基二硫化物与Fe表面2种反应历程的能垒数据Fig.6 Energy barrier data of two reaction processes between diethyl disulfide and Fe surface
通过模拟计算可知,含硫化合物与金属表面的作用机理不是单一的Forbes[4-5]所提出的吸附反应或Najman等[8]所提出的分解反应历程,具体的反应历程会根据工况条件发生变化。同时,Forbes所提出的吸附反应机理,分子与金属表面作用过程中S—S键的断裂是在吸附前还是吸附后难以通过实验确定。通过模拟计算得到,分子与金属Fe表面接近的过程中,S—S键在吸附前就发生断裂,之后单硫烷基与金属Fe表面发生化学作用。由此可见,分子模拟方法能够更深入、更直观地认识含硫化合物与金属表面的作用机理。
2.1.2 二乙基二硫化物与Fe(110)表面的吸附反应过程研究
详细分析二乙基二硫化物与Fe(110)表面的吸附反应历程,作用过程中每步反应的反应热(ΔH)和开始解离/吸附的吸附能(Eads)如图7所示。二乙基二硫化物与Fe(110)表面的吸附能为负值,且绝对值很大,说明二乙基二硫化物能够很稳定地吸附在Fe(110)表面。二乙基二硫化物与Fe(110)表面的解离/吸附过程反应热为负值,说明吸附后产物的能量比初始反应物的能量低,反应为放热过程。之后2个 C—S键断裂的反应热均为正值,说明断裂后产物的能量比反应物能量高,这2个基元反应是吸热过程。

Red number—Reaction heat, kJ/mol; Eads—Adsorption energy, kJ/mol图7 二乙基二硫化物吸附/反应历程中的反应热和吸附能Fig.7 Reaction heat and adsorption energy in the adsorption/reaction process of diethyl disulfide
进一步分析二乙基二硫化物与Fe(110)表面的解离/吸附过程,由图4的构象可知,吸附后二乙基二硫化物中的2个S原子与Fe(110)表面的距离都较小,稳定时每个S原子都与2个Fe原子的距离较近,2个S原子之间的距离较大。分析吸附过程中2个S原子与各自2个Fe原子的距离变化如图8(a)所示,二乙基二硫化物中2个 S原子之间的距离变化如图8(b)所示,将构象中左侧S原子记为S1,右侧S原子记为S2。由图8可以看到:吸附过程中,2个S原子与Fe原子的距离减小,稳定时S1与2个Fe原子的距离为0.2295和0.2245 nm,S2与2个Fe原子的距离为0.2229和0.2311 nm;2个S原子之间的距离增大,稳定时的距离为0.3816 nm。用CASTEP模块计算2个原子之间的键级,得到初始状态下S原子与Fe原子的键级均为0,2个S原子之间的键级为0.420,吸附之后S1与距离较近2个Fe原子的键级为0.530和0.570,S2与距离较近2个Fe原子的键级为0.570和0.470,而2个S原子的键级为0。由此可以说明,二乙基二硫化物与Fe(110)表面吸附过程中,2个S原子与Fe原子作用,并形成化学键,同时Fe原子的作用使分子中的S—S键断裂,这一过程为解离/吸附,吸附能很大,断裂后的单硫化物稳定吸附在Fe(110)表面,这种吸附是化学反应过程。
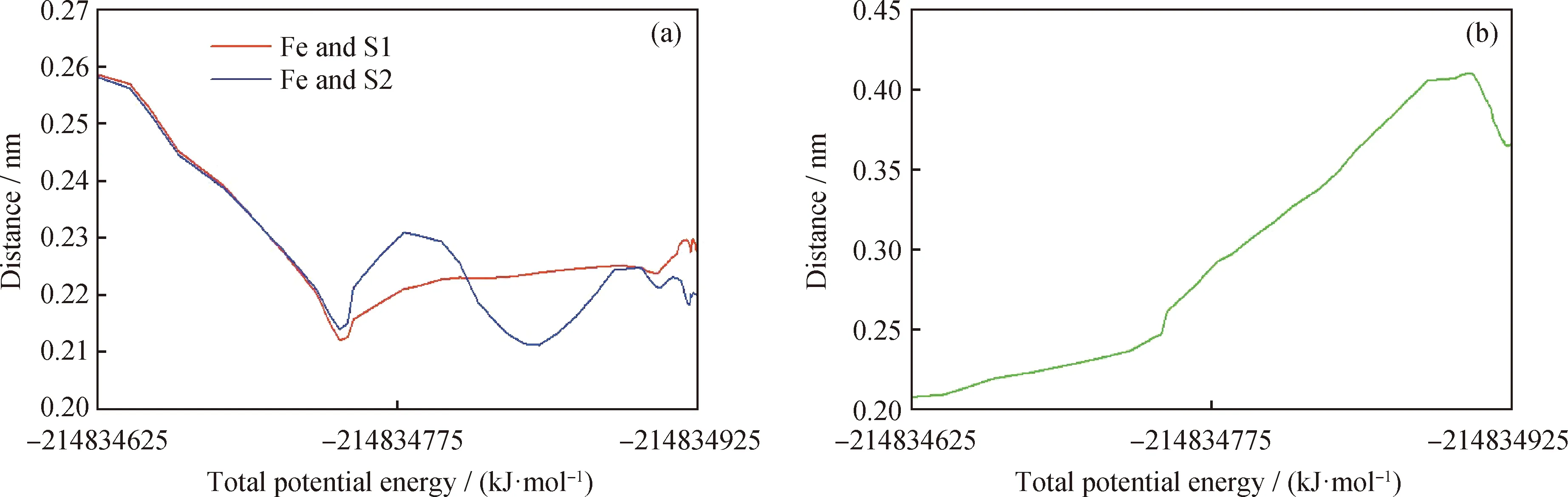
S1—Sulfur on the left in conformation; S2—Sulfur on the right in conformation图8 量子化学模拟二乙基二硫化物与Fe表面作用过程中原子间距离的变化Fig.8 Quantum chemistry simulation of interatomic distance changes during the interaction of diethyl disulfide with Fe surface(a) The distance between Fe and S; (b) The distance between S and S
二乙基二硫化物与Fe(110)表面发生化学吸附的过程中S—S键断裂,单硫化物基团中的S原子与Fe原子形成化学键,之后C—S键断裂,详细研究作用过程中的电子转移及键长、键能、键级变化有助于深入认识其作用机理,吸附反应过程中4种反应物和产物分子构象中S原子的电荷(Q(S))、C—S键键长(D(C—S))、C—S键键能(EBD (C—S))和C—S键键级(B(C—S))如表1所示。

表1 吸附反应历程中二乙基二硫化物分子结构参数的变化Table 1 Changes in the molecular structure parameters of diethyl disulfide during adsorption/reaction
由吸附/反应历程中二乙基二硫化物结构参数的计算结果可知:二乙基二硫化物与Fe(110)表面的解离/吸附过程中,S原子上的电荷减少,S1和S2分别失去0.037和0.071 e的电荷,说明在开始的解离/吸附过程中,二乙基二硫化物提供电子,Fe(110)表面得到电子,作用过程中发生电子的转移,这也可以说明该过程为化学吸附。根据之前分析的硫化异丁烯电子密度分布,分子中2个S原子上的电子密度最大,结合分子通过向Fe(110)表面提供电子与其发生化学作用,则电子密度较大的位点更容易与Fe作用。吸附之后,二硫化物中的2个C—S键键长变长、键能降低、键级变小,说明化学吸附使C—S键活化,更容易断裂,这也可以解释之前吸附在铁表面单硫化物的C—S键断裂能垒较低。
之后1个C—S键断裂,Fe原子容易失去自由电子使S原子带负电,生成FeS,此时另外1个C—S键的各项参数与之前没有明显变化,没有受到较大影响。因此,二乙基二硫化物与Fe(110)表面反应生成FeS的作用过程可描述为:二乙基二硫化物中的S原子通过向Fe(110)表面提供电子发生较强作用的解离/化学吸附,Fe原子与S原子生成化学键;吸附之后单硫化物中的C—S键被活化,键长增加,键能降低,键级变小,更容易断裂,从而生成FeS。
2.1.3 二乙基二硫化物与Fe(110)表面的分解/反应过程研究
分析二乙基二硫化物与Fe(110)表面发生分解/反应历程的反应热,得到二乙基二硫化物中S—S键断裂过程的反应热为270.73 kJ/mol,之后C—S键断裂的反应热为307.03 kJ/mol,这2个过程都是吸热过程,需要外界提供热量,因此要在苛刻条件下发生。之后活性S与Fe(110)表面反应的反应热为-167.99 kJ/mol,该过程是放热反应。
分析作用过程中的S原子上的电荷变化,可以得到,二乙基二硫化物中2个S原子的电荷量分别为-0.012和-0.019 e,S—S键断裂之后,2个单硫化物自由基上S原子的电荷量分别为-0.048和-0.045 e,2个S原子上的电荷量增多,这使得C—S键上的电子密度增加,化学键更难断裂,该步反应需要的热量也更多。活性S与Fe(110)表面反应后,S原子的电荷量为-0.047 e,这是由于Fe原子容易失去电子,使得S原子得到电子,带负电荷。
2.2 含硫极压剂表面膜的性能研究
在温度高于500 ℃条件下,二乙基二硫化物与Fe表面发生化学反应,最后生成FeS膜来发挥抗烧结性能。通过研究1个二乙基二硫化物分子与Fe(110)表面反应后Fe表面的变化,说明FeS膜的生成。二乙基二硫化物中的S1原子与Fe(110)表面最上层原子的Fe1和Fe2结合,具体位置如图9所示,S2原子与Fe(110)表面最上层原子的Fe3和Fe4结合,比较S原子结合前后Fe1、Fe2、Fe3、Fe4原子与周围Fe原子之间的电子重合百分率,用键级表示,结果如图9中的红色数字所示。由图9可以看到:二乙基二硫化物与Fe(110)表面反应之前,Fe1、Fe2、Fe3、Fe4原子与周围原子之间的键级在0.50~0.54之间;反应之后,4个Fe原子与周围原子之间的键级在0.12~0.49之间,原子之间的共用电子数量减少,则Fe原子之间的结合力下降。由前文的分析得到,C—S键断裂后,Fe原子向S原子提供电子,S原子带负电荷,这也说明S原子作用后Fe表面上原子之间的电子减少。因此,硫化物与Fe(110)表面作用后,Fe表面上原子之间的相互作用减弱,更容易断裂。推测在多个硫化物与Fe(110)表面作用后,第一层Fe原子之间的电子密度下降,Fe—Fe断裂,生成FeS,发挥保护金属表面的作用。
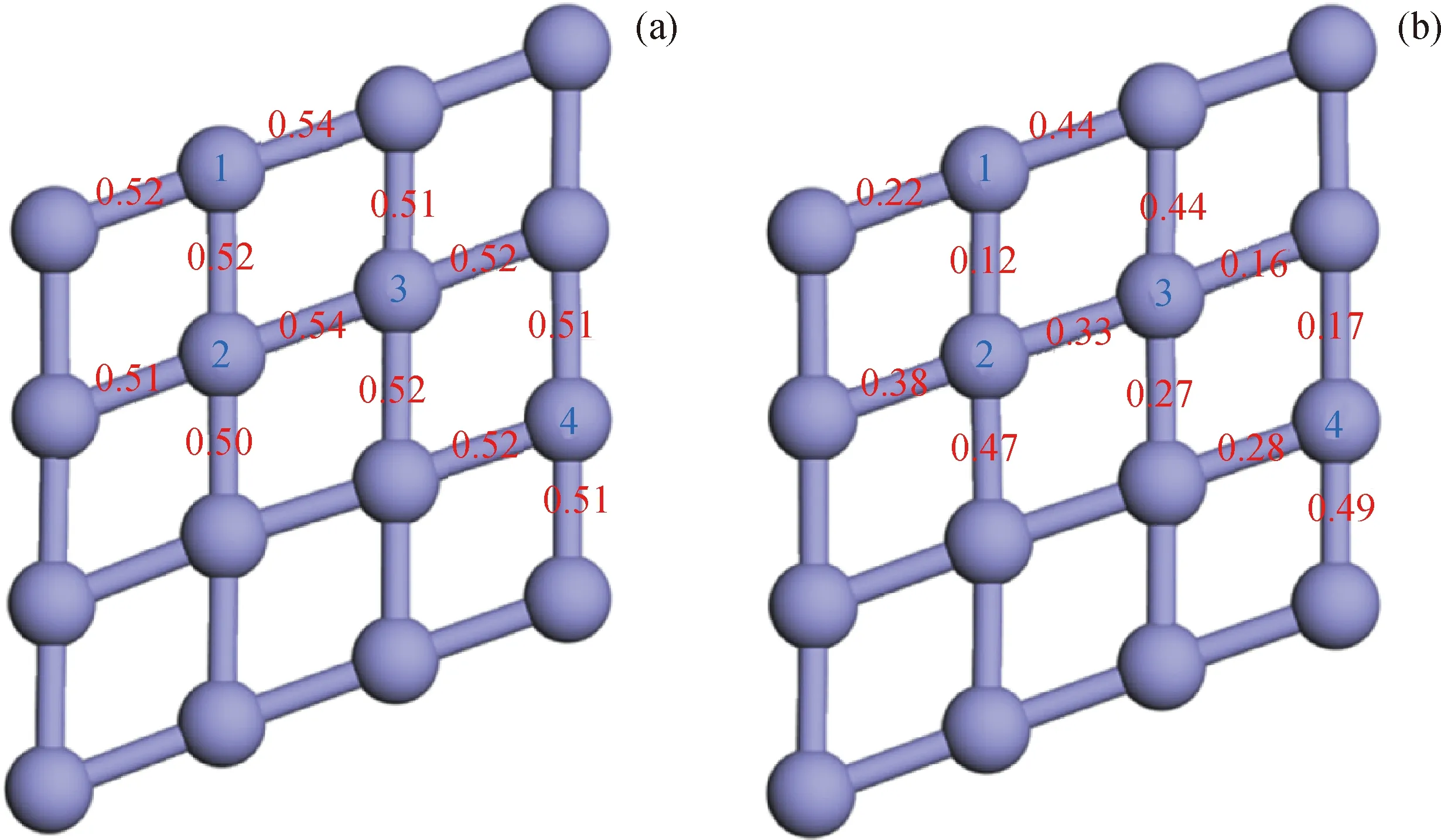
Fe atom; Fe—Fe bond; Red number—Bond order between atoms; Blue number—Mark of iron atom interacting with sulfur atom图9 S原子作用前后Fe(110)表面第一层原子之间的键级Fig.9 Bond orders between the first layer atoms on the surface of Fe(110) before and after S atom interaction with Fe(110) surface(a) Before the reaction; (b) After the reaction
对于FeS表面膜,S原子通过化学键与Fe原子结合吸附在Fe(110)表面,同时组成表面膜的Fe原子和S原子也是通过化学键结合。为研究Fe原子与S原子之间的结合能,用2种FeS晶体为模型化合物,其结构和晶格参数如图10所示。计算这2种FeS晶体内部的原子结合能,同时与纯Fe金属的原子结合能比较,金属Fe的结构如图11所示。量子化学方法计算得到FeS-Ⅰ和FeS-Ⅱ中单位原子之间的结合能为-444.31和-438.62 kJ/mol,金属Fe的单位原子结合能为-525.69 kJ/mol。

Fe atom; S atom; Fe—S bond图10 FeS的晶体结构和晶格参数Fig.10 Crystal structure and lattice parameters of FeS
Fe atom; Fe—Fe bond图11 Fe的晶体结构Fig.11 Crystal structure of Fe
FeS层中,Fe与S原子间以键合作用结合,结合能很大,能够在高温下存在于Fe表面。高温下Fe表面的烧结主要是因为2个金属表面的Fe原子键合在一起,结合能很大,表面的剪切运动已不足以克服Fe原子的结合力,从而发生金属部件的卡咬。当在Fe表面上生成FeS层时,表面相对运动要克服的阻力是FeS层内Fe原子与S原子间的键合作用能,该值比Fe原子间的低,更容易发生相对运动,避免了Fe表面的卡咬,因此FeS膜能发挥抗烧结作用。同时FeS层被剪切破坏后,硫化物又很容易与Fe表面反应继续生成FeS,从而持续发挥保护抗烧结作用。
由上述分析可知,含硫化合物形成原子间键合作用很强的FeS无机物膜,能够在苛刻条件下发挥抗烧结的作用。同时,润滑油组分形成表面保护膜的内聚能或原子间结合能应比Fe表面小,这样在剪切运动时,磨损优先发生在作用力较弱的位置,即先磨损表面保护膜,油中的组分继续生成表面保护膜,从而避免金属表面的磨损和烧结。
3 结 论
(1)边界润滑状态下,工况较温和时,二硫化物更容易先与Fe表面发生不可逆的化学吸附,S—S在吸附时先断裂,进而C—S键断裂,生成FeS,以吸附反应历程为主;工况较苛刻时,能够使二硫化物中的S—S键、C—S键断裂,则生成的活性硫很容易与Fe表面反应,生成FeS,即在高温下,以分解反应历程为主。
(2)FeS膜内部主要是化学键合作用,其键合能很大,能够在高温下存在于Fe表面;同时FeS膜内原子间的键合能比Fe原子间的低,表面的剪切运动主要克服Fe和S原子相对较弱的键合作用,而非键合能很强的Fe和Fe原子间作用,一定的剪切作用使表面可以做相对运动,从而避免了Fe表面的卡咬,发挥抗烧结作用。
(3)由于硫化物与Fe表面的化学作用很强,在FeS消耗后,又容易继续生成新的FeS,从而在高温下持续发挥抗烧结作用。
