FLPs催化烯胺还原活性研究及反应机理探讨
2022-11-15温志国谯在银田冲MaximBorzov聂万丽
温志国,谯在银,田冲,Maxim Borzov,聂万丽
(1.乐山师范学院新能源材料与化学学院,天然产物化学与小分子催化四川省高校重点实验室,乐山 614000;2.峨眉山宏昇药业股份有限公司,峨眉山 614200)
2006年,Stephan课题组[1]在《Science》期刊上发表了第一篇有关受限路易斯酸碱对(Frustrated Lewis pairs,FLPs)对小分子活化的研究工作.研究发现了位阻较大的路易斯酸碱对之间通过相互协同作用在温和条件下对H2的活化反应特征,使得人们对主族元素在活化H2和还原氢化反应中的应用产生了极大的兴趣.在过去的十几年间,由各类大位阻路易斯酸(Lewis acid,LA,如硼烷)及其与大位阻路易斯碱(Lewis base,LB,如有机膦和有机胺)所组成的结构各异的FLPs体系,被用于对H2、CO2、烯烃、炔烃、环氧化物、亚胺、羰基化合物、烯醇硅醚和芳香杂环等的活化、转化及选择性还原,为基于硼烷的非金属催化化学开辟了更广阔的应用前景(图1)[1~10].

Fig.1 Chemical reactivities of FLPs
2014年,Ingleson等[11]发现,在弱碱作用下,B(C6F5)3可催化还原烯胺,生成氢化硅烷加成产物(图2).按照常规FLPs催化的烯胺硅氢化加成反应机理,尽管会产生一定量的氢化还原产物,但转化率最高不超过50%.其加成反应机理如图2所示.
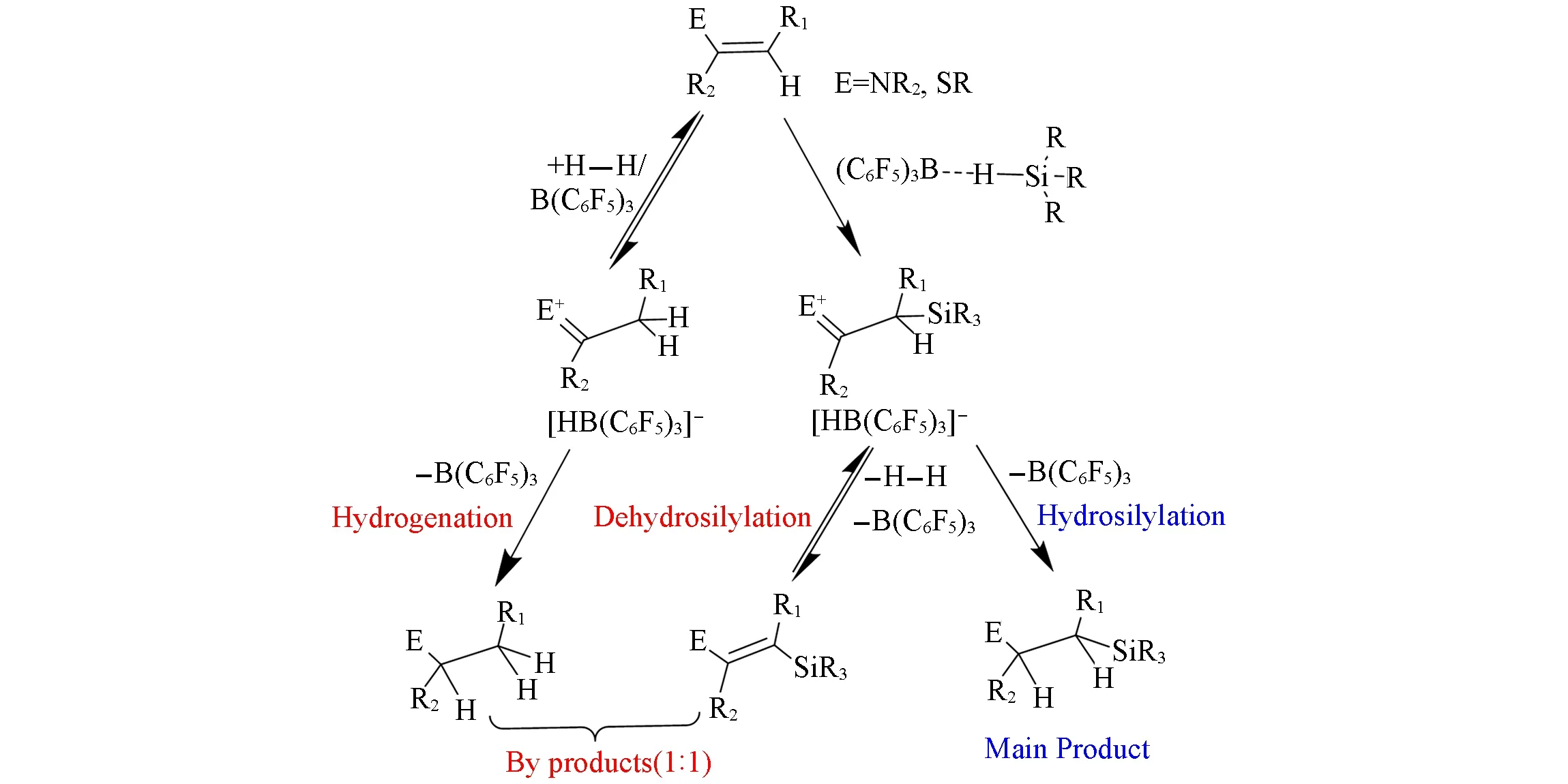
Fig.2 Dehydro-/hydro-silylation and hydrogenation of enamines with borane
对于FLPs活化小分子的机理,目前文献报道主要有2种观点.有关FLPs对氢气的活化,最早的报道[12,13]认为H—H键在FLPs催化异裂过程中首先形成一种线型中间体,如t-Bu3P…H…H…B(C6F5)3.随后Rieger等[14]认为FLPs对H2等小分子的活化机理可能是通过一种非线型的中间过渡态(图3).在该中间体中,由于空间拥挤,路易斯酸碱中心相互靠近但未成键,在FLPs中间形成一个可以被小分子X2插入的通道.X2分子在供电子原子LB与受电子原子LA的静电场的作用下被极化,发生异裂[13,15].尽管这2种活化机理不同,但相同点是均认为FLPs是通过催化异裂X—X键的过程对小分子进行活化的.目前文献中有关FLPs催化活化的反应多采用此异裂方式对反应机理进行研究讨论.
2017年,Stephan课题组[16,17]对FLPs化学活性的研究中发现了一种不同于传统的催化异裂X—X键的机制,认为受限路易斯酸碱对通过单电子的转移,可形成受限自由基离子对(FRPs)形式的活性中间体.这种FRPs可以通过催化均裂X—X键的方式对小分子进行活化(图3).
本课题组[18~23]对以氢化硅烷作为还原剂,B(C6F5)3作为路易斯酸的FLPs的催化体系进行了系统研究.在该体系催化烯胺类底物的还原反应中发现,反应并未按照传统FLPs催化异裂的方式得到预期的硅氢化产物,而是主要得到烯胺的氢化还原产物(图4).本文对不同的路易斯碱、不同取代的氢化硅烷以及不同结构的烯胺进行研究发现,无论在何种光照条件下,硅氢化反应都不是主要反应.在此基础上,本文利用氘代硅烷试剂对反应机理进行了相应的同位素效应研究.
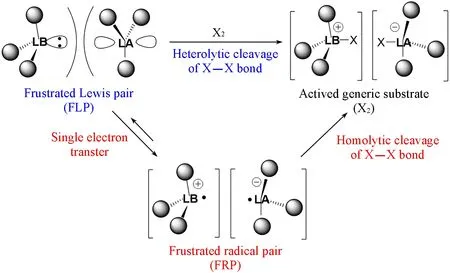
Fig.3 Mechanisms of small-molecule activation by FLPs

Fig.4 Reduction of enamines catalyzed by FLPs
1 实验部分
1.1 试剂与仪器
1-(1-环己烯基)哌啶(纯度97%)、1-(1-环己烯基)四氢吡咯(纯度97%)、4-(1-环己烯基)吗啡啉(纯度97%)、N-(1-环戊烯基)吗啡啉(纯度97%)、苯硅烷(PhSiH3,纯度98%)、二苯基硅烷(Ph2SiH2,纯度99%)、氘代二苯基硅烷(Ph2SiD2,纯度99%)、三苯基硅烷(Ph3SiH,纯度99%)、三乙基硅烷(Et3SiH,纯度99%)、三异丙基硅烷(i-Pr3SiH,纯度97.5%)、三五氟苯基硼[B(C6F5)3,纯度97%]、2,2,6,6-四甲基哌啶(TMP,纯度98%)、三叔丁基膦(t-Bu3P,纯度98%)、三异丙叉丙酮基膦(MeS3P,纯度98%)和三苯基甲烷(Ph3CH,纯度99%),北京百灵威科技有限公司.
Avance III 400 MHz型核磁共振波谱仪(NMR),瑞士Bruker公司;LE104E型电子分析天平,美国梅特勒公司.
1.2 实验过程
1.2.1 不同因素对烯胺还原活性的影响在核磁管中加入5.1 mg(0.01 mmol)B(C6F5)3和0.01 mmol LB(tBu3P,TMP,MeS3P),加入适量CDCl3溶解,然后加入15.3 mg(0.01 mmol)N-(1-环戊烯基)吗啡啉,最后加入一定量的氢化硅烷作为还原剂进行反应;反应完成后进行1H NMR测试,计算产率.
1.2.2 不同烯胺的反应活性研究在3根核磁管中各加入5.1 mg(0.01 mmol)B(C6F5)3,3.9 mg(0.01 mmol)MeS3P,36.9 mg(0.2 mmol)Ph2SiH2和9.8 mg(0.04 mmol)Ph3Ch,加入适量氘代氯仿溶解;再分别加入16.7 mg(0.1 mmol)4-(1-环己烯基)吗啡啉、15.1 mg(0.1 mmol)1-(1-环己烯基)四氢吡咯和16.5 mg(0.1 mmol)1-(1-环己烯基)哌啶,在365 nm波长光照下反应6 h.反应完成后进行1H NMR测试,计算产率.
1.2.3 同位素跟踪实验在3根核磁管中各加入5.1 mg(0.01 mmol)B(C6F5)3,3.9 mg(0.01 mmol)MeS3P,36.9 mg(0.2 mmol)Ph2SiD2和9.8 mg(0.04 mmol)Ph3Ch,加入适量氘代氯仿溶解;再分别加入16.7 mg(0.1 mmol)4-(1-环己烯基)吗啡啉、15.1 mg(0.1 mmol)1-(1-环己烯基)四氢吡咯和16.5 mg(0.1 mmol)1-(1-环己烯基)哌啶,在365 nm波长光照下反应6 h.反应完成后进行1H NMR测试,计算产率.
另取3根核磁管,各加入5.1 mg(0.01 mmol)B(C6F5)3,36.9 mg(0.2 mmol)Ph2SiD2,15.3 mg(0.1 mmol)N-(1-环戊烯基)吗啡啉和9.8 mg(0.04 mmol)Ph3Ch,加入适量氘代氯仿溶解;再分别加入1.4 mg(0.01 mmol)TMP,10 μL(0.01 mmol)tBu3P和3.9 mg(0.01 mmol)MeS3P,在365 nm波长光照下反应6 h.反应完成后进行1H NMR测试,计算产率.
2 结果与讨论
2.1 对烯胺还原反应活性影响因素的探讨
首先研究了不同取代的还原剂R3SiH、不同路易斯碱LB、反应时间及不同光照条件对N-(1-环戊烯基)吗啡啉(1a)的还原反应活性的影响,所得数据列于表1.
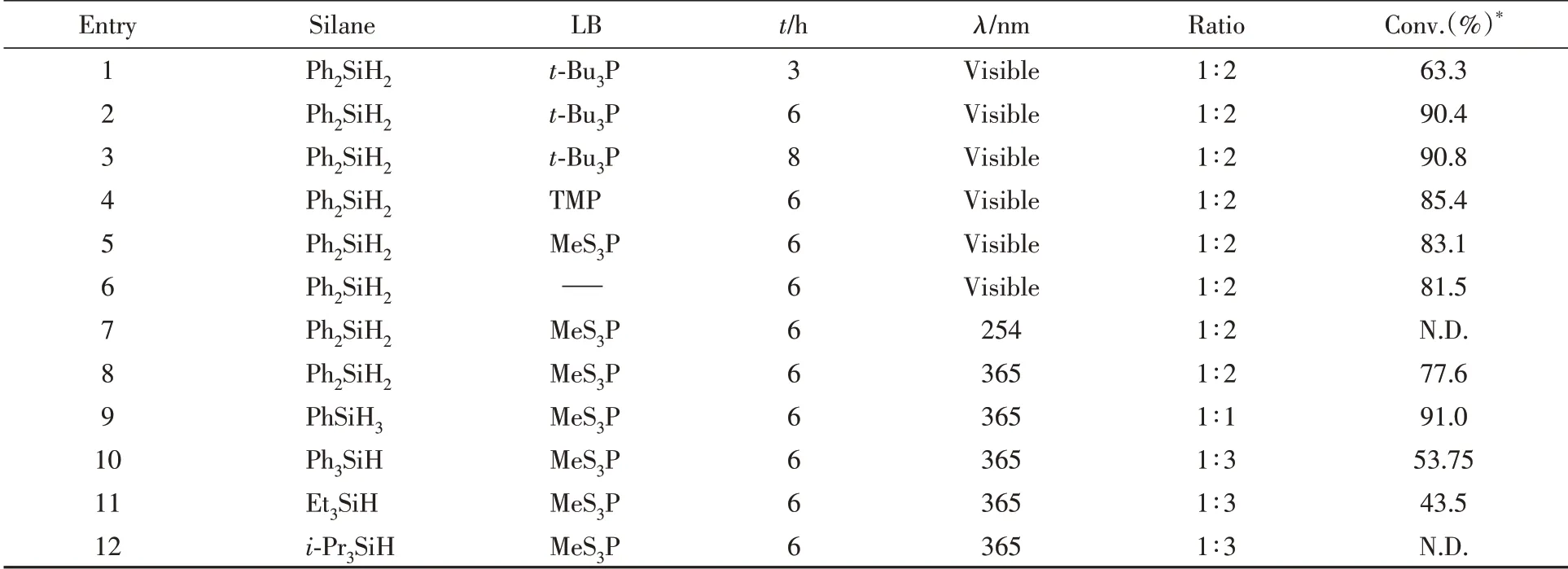
Table 1 Screening reaction conditions
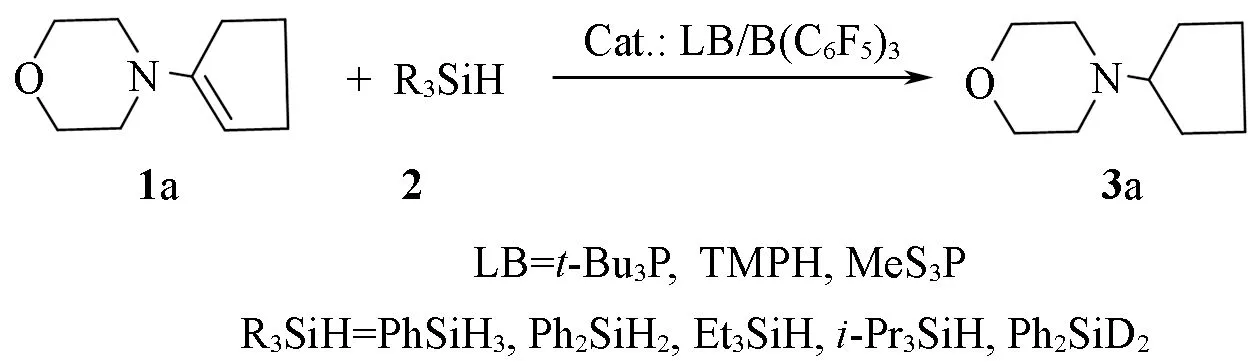
*The conversion of reduced product 3a was analyzed by NMR with Ph3CH as internal standard.
从表1中Entries 1~3可以看出,以Ph2SiH2为还原剂,以B(C6F5)3与t-Bu3P组成的FLP为催化体系,在可见光光照下6 h后,化合物1a转化为化合物3a的转化率高达90%以上,远高于光照3 h的反应结果.光照8 h对转化率提高不明显.而B(C6F5)3与TMP和MeS3P分别组成的FLPs催化体系(表1中Entries 4和5),在6 h可见光光照下转化率也达到80%以上.表1中Entry 6数据表明,B(C6F5)3单独催化的转化率比有LB参与下的相对低一些.
从表1中Entries 7和8可以看出,在反应条件相同的情况下,以B(C6F5)3与MeS3P组成的FLP为催化体系,不同的光照条件具有不同的反应活性:在254 nm光照条件下几乎不反应,而在365 nm光照条件下的转化率77.6%,明显低于可见光光照下的83.1%.表1中Entries 8~12数据显示了在365 nm光照条件下,同样以B(C6F5)3与MeS3P组成的FLP为催化体系,不同氢化硅烷对化合物1a的还原活性的影响.可以看出,芳基取代硅烷的活性大于烷基取代硅烷,且随着取代基的增加,活性降低.对于三烷基取代的硅烷,取代基的空间位阻会使硅烷的活性降低,三异丙基取代的i-Pr3SiH不能还原化合物1a.从表1数据还可以看出,活性最高的PhSiH3,在365 nm光照条件下对化合物1a的催化还原活性最高,转化率可高达91.0%,高活性的芳香性氢化硅烷在365 nm或可见光光照条件下,其相应化合物3a的转化率都可达到50%以上.所有反应中均未观察到硅氢化加成产物生成.
2.2 不同烯胺的反应性及还原机理探讨
在考察了影响烯胺还原反应活性的因素之后,研究了一定条件下不同取代的烯胺的还原反应活性.以B(C6F5)3与MeS3P组成的FLP为催化体系,Ph2SiH2为还原剂,在365 nm光照条件下,对3种烯胺化合物1b~1d进行还原,转化率见表2中Entries 1~3.可以看出,虽然其转化率相对于化合物1a都不高,但均达到50%以上.
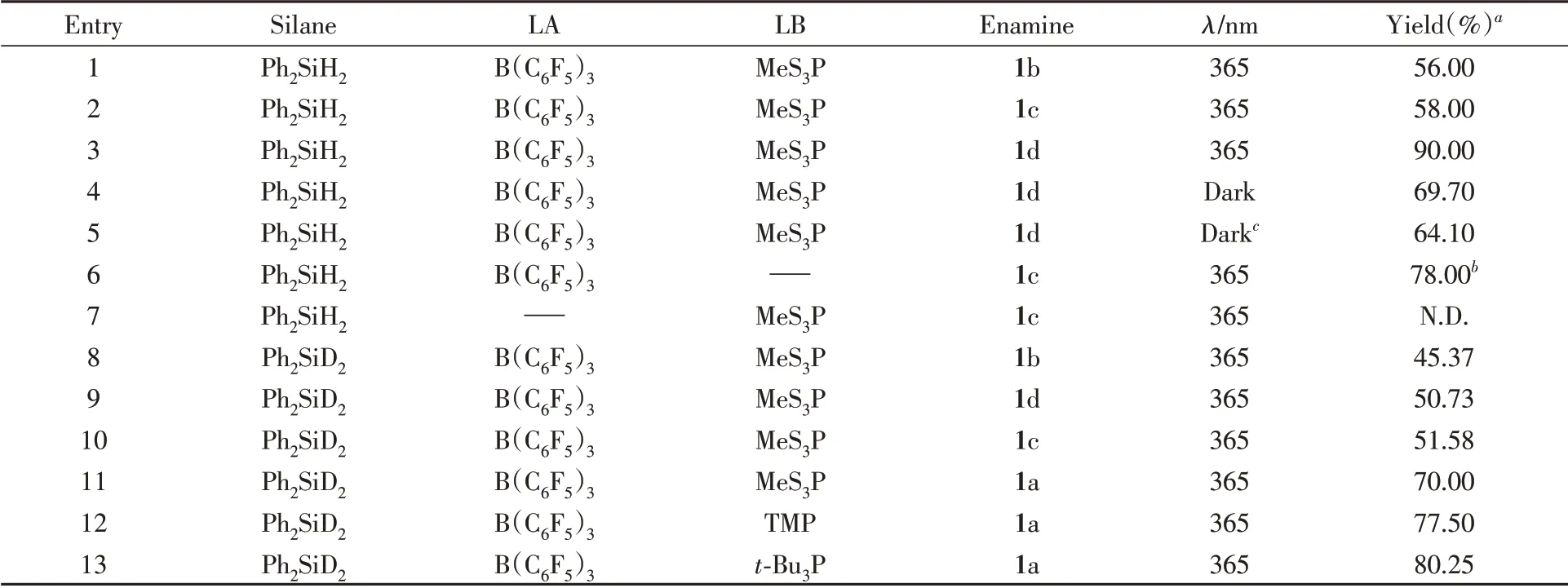
Table 2 Scope of reduction and isotope effect

a.The yield of product 3 were analyzed by NMR with Ph3CH as internal standard;b.the conversion rate was calculated corresponding to the enamines;c.the reaction temperature was 50℃.
比较表2中Entries 3和4可以看出,光照条件下反应的产率高于避光条件.从表2 Entries 4和5可以看出,在避光条件下,温度对反应的影响不大,说明反应体系可能易产生自由基,但由于自由基存在时间短,以2,2,6,6-四甲基哌啶氧化物(TEMPO)为自由基捕获剂研究捕获产物时,也未观察到自由基中间体产物.表2中Entries 6和7数据表明,B(C6F5)3是反应必不可少的Lewis酸催化剂,其原因可能是原料烯胺本身就是一种Lewis碱,可以与硼烷之间进行单电子转移形成活性自由基,使反应得以发生,因此反应体系中不加入MeS3P时反应也可以进行.
为了对反应的机理进行深入探究,选择以氘代硅烷Ph2SiD2进行同位素效应研究.从表2中Entries 8~13可以看出,在相同的底物和反应条件下,Ph2SiD2所还原产物的转化率均有不同程度的降低现象,因此推测在反应的过程中,氢化硅烷应该参与了决速反应步骤.
图5为分别在Ph2SiH2及Ph2SiD2的还原下,烯胺1d的转化产物3d和3d′的核磁共振氢谱.可以观察到使用氘代还原剂的条件下,环己烷环上的N—CH质子信号峰在对应位置消失,同时杂环上与氮原子相邻的N—CH2信号峰受到部分氘代的影响而变矮,变得不够尖锐.
图6为Ph2SiD2还原烯胺1d的核磁共振碳谱.由于受到D的自旋效应的影响,在δ63.5处的三重峰(1∶1∶1)对应于环己烷环上的N—CD化学位移,同时杂环上与氮原子相邻的N—C信号峰由于受到部分氘代的影响而变矮,其高场附近也可观察到对应(1∶1∶1)三重峰.
由图5和图6可见,氘代硅烷(Ph2SiD2)的D并未直接还原C=C双键[图7(A)],而是发生了比较复杂的迁移现象,2个D分别连接在氮原子的邻位叔碳CH—N及一个仲碳CH2—N上[图7(B)].
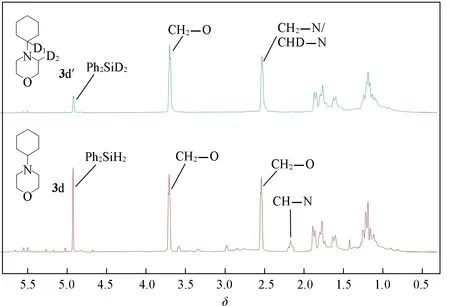
Fig.5 1H NMR spectrum of the deuterated products 3d and 3d′

Fig.6 13C NMR spectrum of the deuterated product 3d′
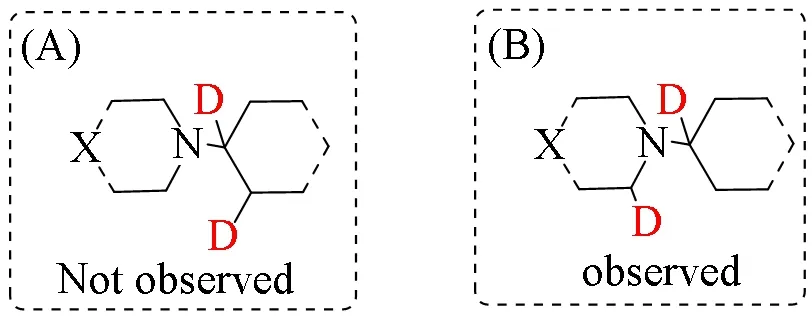
Fig.7 Plausible deuterated reduction products

Fig.8 Speculated mechanism of enamine reduction catalyzed by FLPs
结合表1及表2数据及核磁共振波谱,推测该类反应可能是由受限自由基对FRP机制催化诱导的自由基参与的反应.如图8所示,首先,反应体系中的路易斯酸碱对(也可以是原料烯胺与硼烷)之间进行单电子转移形成活性FRP(i),同时烯胺通过共振化形成的亚胺盐可促使烯胺的分子内氢迁移(ii),氮原子邻位的质子迁移到双键碳上的同时,路易斯碱自由基阳离子可获取1个电子而被还原(iii),并形成氮杂烯丙基自由基或阳离子;另外,路易斯酸自由基阴离子分别与2分子氢化硅烷进行自由基反应,形成D—B(C6F5)3阴离子,2分子硅烷自由基可结合成相应的二硅基化合物(iv);最后,D—B(C6F5)3阴离子进而还原相应的氮杂烯丙基自由基或阳离子成胺(v).
3 结论
对系列受限路易斯酸碱对催化的氢化硅烷还原烯胺进行了研究.结果表明,按照传统FLPs催化异裂的方式不能对产物的氢化还原现象进行解释;结合同位素效应的研究,推测反应按照自由基机理进行的可能性最为合理.尽管有关FLPs化学的发展已经有近15年的研究背景,但其催化反应的机理尚有未知的可能性需要进一步的探索.目前,相关机理的理论计算化学研究正在进行中,本文研究结果不仅对探索FLPs体系的催化方式而且对进一步拓其应用领域具有重要的意义.