过渡金属磷化物析氢催化剂的掺杂调控
2022-11-15王祖民孟程于然波
王祖民,孟程,于然波,3
(1.北京科技大学冶金与生态工程学院,储能科学与工程系,北京 100083;2.中国科学院过程工程研究所生化工程国家重点实验室,北京 100190;3.郑州大学材料成型及模具技术教育部重点实验室,郑州 450002)
一直以来,化石燃料作为主要的能源载体,满足了人们工商业的发展.全球消耗的85.3%以上的能源由不可再生的石油、天然气和煤炭供应[1].但是巨大的消耗量产生了严重的环境污染问题,如酸雨、有害尘埃颗粒及温室气体大量排放造成全球变暖.同时,随着工商业的进步,能源大量使用造成的短缺问题是遏制发展的重要因素.为了满足工商业巨大的能源供应需求,科研人员开始对太阳能、风能、氢能等进行深入广泛的研究.
氢气能量密度高、燃烧产物无污染,有希望作为化石燃料的替代品,因此被称为终极能源[2].然而氢气不是天然存在的,通常需要从含氢原料中制备.化石燃料(如甲烷和煤炭)重整制氢技术应用广泛,但是存在制备得到的氢气纯度低和生产过程排放大量温室气体的问题.新兴的光催化技术是利用光辐射的能量使半导体材料内的电子和空穴发生分离[3],然后将水还原成氢气或氧化成氧气,是一种节能且环保的制氢手段,但是其效率极低,无法满足生产需求[4,5].水电解法通过接入氯碱工业设施可以制备出高纯度的氢气,是一种很有发展前景的清洁化学燃料生成技术.
当在阴极和阳极两极加上外电压后,可分别析出氢气和氧气,总反应为

其中,析氢反应过程的第一步称为沃尔默反应(Volmer reaction),根据电解质的不同表现为:在酸性溶液中,一个迁移到电极表面的电子还原一个质子H+生成氢中间体(H*):

而在中性/碱性溶液中,需要通过电子迁移还原水分子来产生H*:

在随后生成氢气的过程中,由于催化剂表面的氢中间体(H*)的覆盖程度不同,也可分成两种情况.如果H*覆盖率比较低,H*就与催化剂上的另一个被电子还原生成的H*结合形成氢气分子,这个过程称为海洛夫斯基Heyrovsky反应.
在酸性溶液中,因为H+含量很高,所以H*就和另一个H+结合生成H2:

因为中性/碱性溶液中的H+含量低,则发生如下反应:

但如果催化剂表面氢中间体(H*)的覆盖程度高,在此情况下相邻的两个H*将直接在催化剂表面结合生成一个H2分子,这个过程称为塔菲尔(Tafel)反应:

由上述分析可知,无论何种反应溶液体系,电催化析氢过程都相对简单,效率较高.然而,目前仅有4%的氢气是通过电解水的方法来制备[6],电解槽成本过高和能耗较大是制约电解水发展的主要阻碍之一.电解水的理论分解电压是1.23 V,然而电解过程需要克服阳极和阴极内在固有活化能垒所需的过电位(ηa,ηc)和补偿系统内阻所需的过电位(ηΩ),往往需要额外的极化过电位(η)才能驱动整体的水分解过程.为了降低电解成本,就需要开发活性高、运行稳定良好的催化剂尽可能地减小电解过电位,提高析氢和析氧的反应速率.贵金属铂(Pt)和二氧化钌(RuO2)等被认为是有效的电解水催化析氢和析氧的催化剂,但是铂族贵金属价格昂贵并且储量稀少,不能被广泛应用于各种电解槽设备,满足大规模安全可靠制氢的需求.因此,研究制备活性高、稳定性好并且成本低廉的电解水催化剂,仍是科研人员面临的重大难题.
过渡金属W,Mo,Co,Cu,Ni及Fe等地壳丰度高,且具有丰富的孤对电子和未满d轨道,有利于通过改变电子态密度、移动d带中心和调整电子自旋态来提高其本征催化活性[7].过渡金属与非金属形成的氮化物(Transition metal nitrides,TMNs)、硫化物(Transition metal dichalcogenides,TMDs)、硼化物(Transition metal borides,TMBs)、碳化物(Transition metal carbides,TMCs)和磷化物(Transition metal phosphides,TMPs)等已被证明是有前途的电解水催化剂[8~10].
磷化物因具有合成方法简单、种类丰富可调、催化高效稳定的特点而被广泛研究[11~13].在此之前,Xiao等[14]系统总结了过渡金属化合物的各种合成方法,着重分析了磷源种类、磷化方式等磷化条件对析氢反应性能的影响.Hu等[15]以磷化镍为切入点,详细介绍了液相合成、次磷酸盐热磷化、红磷热磷化、磷酸盐氢还原和电化学沉积法5种制备磷化物的方法,并分别讨论了它们的优缺点:次磷酸盐或红磷热磷化法制备的磷化物表现出更高的电催化析氢活性,并有利于保持其前驱体形貌;溶液相法适用于制备磷化镍催化剂油墨,与印刷技术生产膜电极兼容有利于大规模应用.Ge等[16]则重点关注了Fe,Co和Ni基磷化物的合成,从前驱体制备(水热反应、静电沉积和湿化学方法)和磷化处理(气固反应、液相反应和电化学还原)两个方面进行了深入分析讨论,对比了各种合成方法的优劣.Wehrspohn等[17]讨论了具有异质结结构的过渡金属磷化物,并分析了其增加活性中心、加速的质量/电荷转移和优化的中间体吸附的协同作用,以及其作为析氢(Hydrogen evolution reaction,HER)/析氧(Oxygen evolution reaction,OER)双功能催化剂的独特优势.Selomulya等[18]则回顾了近些年的研究进展,提出了包括调整M/P(M=W,Mo,Co,Cu,Ni,Fe)比、多金属合金化、与碳材料杂化复合、表界面工程、构建多孔结构等提升过渡金属磷化物电解水催化性能的几大策略.Ray等[19]关注了特定的磷化镍材料,总结了通过不同的制备方法和处理手段,改变其相、尺寸、形貌等因素,来调控电解水性能.目前文献报道主要集中在材料合成和微纳结构设计方面,对其电子结构的调控以提高固有催化活性等相关内容没有深入的探论.
本文尝试根据最新的理论和实验成果,从元素掺杂实现电子结构调控的角度为设计高活性的HER催化剂提供一些建议.首先讨论了TMP作为HER催化剂的的特点和不足,接着从实验和密度泛函理论(DFT)两方面分析了物理化学性质不同的各种金属、非金属掺杂元素对磷化物电子结构的影响,并讨论这些电子结构的调整如何在催化剂上激活或创造活性位点、调解氢吸附强度、促进水的解离,最后建立了掺杂造成的电子结构变化与催化活性优化的关系,并对未来的研究方向进行了展望.
1 过渡金属磷化物
2005年,Liu等[20]首先发现了TMPs作为电催化剂的潜在优势,Ni2P(001)晶面表现出类似氢化酶的性质.金属位点带部分正电荷和P位点带部分负电荷分别作为氢化物受体和质子受体,金属与非金属之间的“集合效应”(Ensemble effect)共同促进HER反应,甚至表现出媲美于Pt的HER活性[21].之后,报道了许多表现出优异HER催化性能的磷化物,如FeP[22,23],FeP3[24],CoP[25~27],Ni2P[28~30],Cu3P[31],MoP[32]等.这些研究表明,良好的性能来源于磷化物独特的结构:在形成晶体时,由于磷原子的半径较大,一般采取占据结构单元内部空间的填充方式,形成的最小结构单元为三棱柱,而这些三棱柱通过不同组合可形成不同的晶格类型,均可以暴露更多不饱和的活性位点.同时P占据三棱柱的内部空隙,具有更高的稳定性.另一方面,磷化物中不仅含有金属-磷键(M—P),还存在大量磷-磷键(P—P),金属-金属键(M—M),这使得过渡金属磷化物具有类似合金的性质,导电性能较强[16].
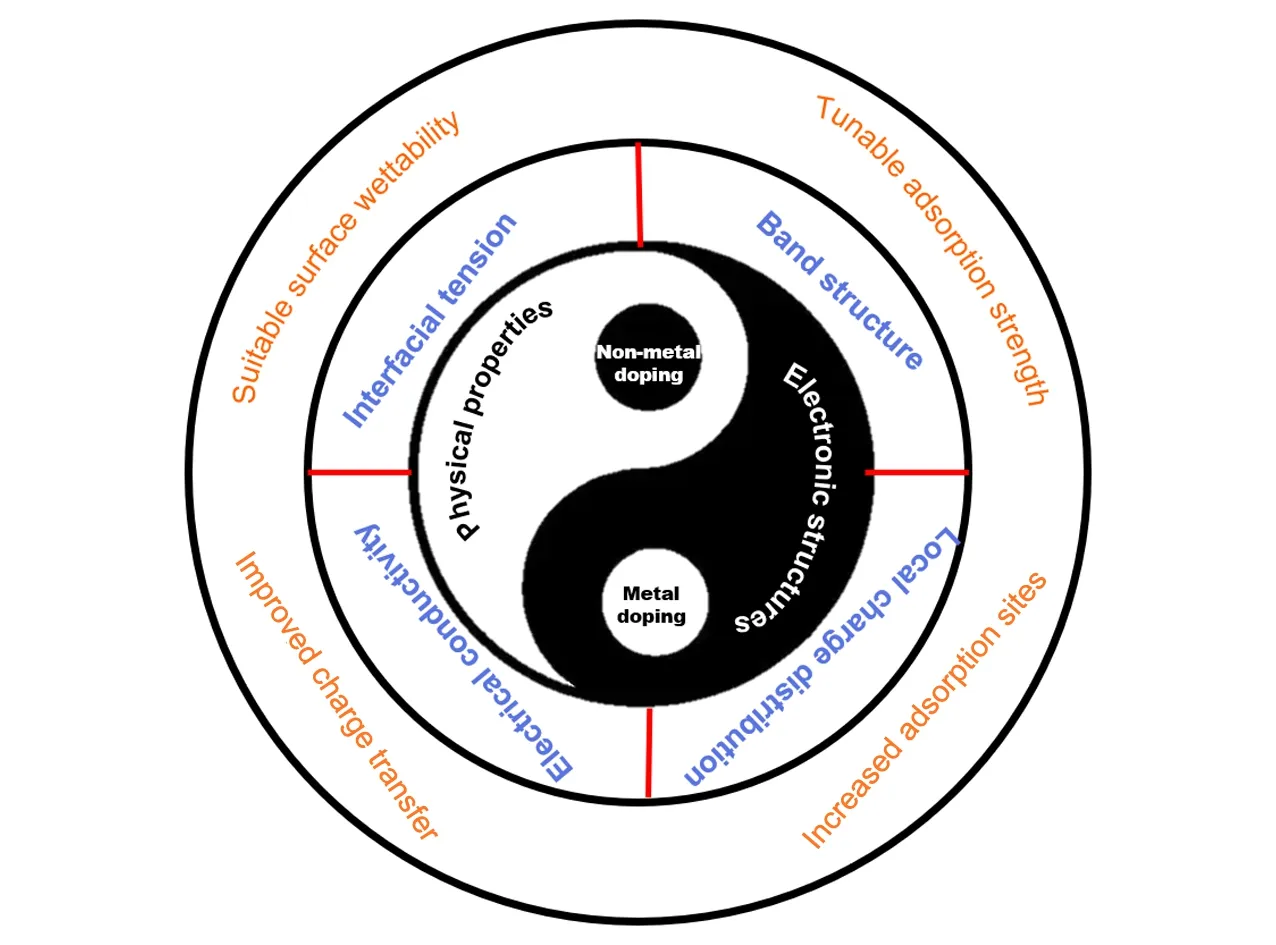
Fig.1 Schematic illustration of the effects of doping regulation in transition metal phosphides
虽然单金属的TMPs在电催化产氢方面表现出了令人惊喜的催化能力[25],但由于其传质效率低、氢中间体不易吸附、脱附等因素,其实际性能仍无法与铂基催化剂相媲美[16].目前研究工作主要集中在以下3个方面来进一步提高过渡金属磷化物的催化活性(图1):(1)结构工程.通过腐蚀等方法合理地去除前驱体中的某一组分,构建多孔结构,或构建层次化的多级结构,使其具有更大的电化学活性面积[33];(2)构建异质多相结构.利用两相间强的电子相互作用,诱导界面处电子重新排布,调整界面电子结构,优化相界面处的吸附能,从而促进H的吸附和脱附[17];(3)掺杂改性.通过掺杂电负性不同的金属或非金属元素来调节磷化物主相的电子结构,从而增强活性位点的内在活性或提供额外的活性位点,优化氢吸附吉布斯自由能(ΔGH*)和水吸附吉布斯自由能(ΔGH2O*),使其易于吸附和解吸,从而表现出比单金属磷化物更好的催化活性[34,35].
掺杂作为最有效和直接调节材料电子结构的方式,将影响反应物产物在催化剂表面活性位上的吸附、解离、脱附等过程,优化每个活性位点的本征活性.众所周知,HER过程中的限速步骤与H*和催化剂表面之间的亲和力有关,当催化剂与H*的结合强度较弱,则Volmer步骤主导HER过程,Tafel斜率约为120 mV/dec;结合力过强时,则Heyrovsky步骤或Tafel步骤将是限速步骤,Tafel斜率分别是40和30 mV/dec[36].根据Sabatier原理[37],高效的催化反应进行需要适中的吸附强度既可以促进H*的形成,又可以容易地解吸形成的分子H2.因此,将ΔGH*作为密度泛函计算DFT中吸附强度的一个描述符,如果氢中间体H*的ΔGH*接近于热中性(0 eV),则H*的吸附强度是最优的,设计的催化剂预计具有高性能[38].
Nørskov等[39]通过实验和DFT方法研究了过渡金属和贵金属的HER活性,最终获得了一个火山图,其中交换电流密度值(j0)是ΔGH*的函数,贵金属Pt的ΔGH*最接近于0,位于火山图的峰顶,对应最高的催化性能,如图2(A)所示.Jaramillo等[40]从理论和实验上发现单金属磷化物的ΔGH*值一般较负或较正,说明氢结合强度太强或太弱.如图2(B)和(C)所示,对比一系列掺杂和未掺杂的TMPs发现,双金属磷化物Fe0.5Co0.5P具有接近热中性的ΔGH*和良好的HER活性,显示出归一化到电化学活性面积(ECSA)时最高的电流密度.该结果成功地证明了材料表面的电子结构决定了ΔGH*值[41],通过引入杂原子可以改变催化剂的局部电子结构,从而达到调节d带中心,优化氢中间体吸附强度的目的.此外,还可以降低电荷转移电阻、提升催化剂活性.
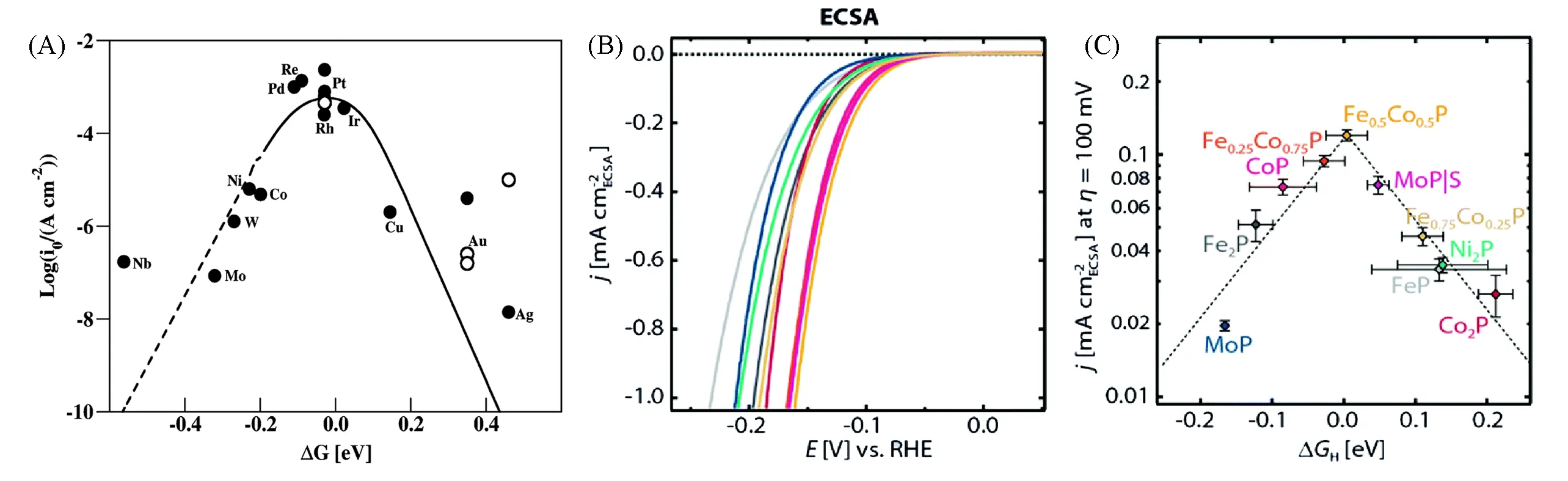
Fig.2 A calculated volcano plot(A)[39],the LSVs normalized to the electrochemical active surface area(ECSA)(B),activity volcano for the HER showing the ECSA normalized current density at η=100 mV as a function ofΔGH(C)[40](A)Copyright 2010,American Chemical Society;(B,C)Copyright 2015,Royal Society of Chemistry.
2 金属掺杂
2.1 单金属掺杂
催化剂的表面电子结构(如d带中心和电子分布)是决定活性的重要因素.许多金属杂质被证明是TMPs的电催化活性促进剂,这归因于它们能够改变主体元素的电子结构,从而增强本征活性.引入杂原子后,由于原子之间的协同效应,会导致主体金属磷化物中的电荷重新分布,对ΔGH*进行调制,从而促进HER,通常表现出比未掺杂的单金属磷化物催化剂有更好的催化活性[42].
Zhang等[43]的研究表明,即使是微量掺杂也会对TMP催化性能产生重大影响.仅仅掺杂0.02%或0.05%(质量分数)Ni或Co的MoP均表现出显著加速的反应动力学.特别是,0.02Ni-MoP在10 mA/cm2电流密度下,相较原始MoP的过电位下降108 mV,他们通过密度泛函理论研究证明,在MoP中微量掺杂镍或钴会导致电荷从掺杂元素转移到主体磷化物基质,调制了H的吸附能,优化了HER的性能.为了了解在碱性介质中金属掺杂对磷化物催化析氢活性的影响,Pan等[44]以ZIF-67为模板,采用自模板转化法合成了过渡金属M掺杂的CoP(M=Ni,Mn,Fe)空心多面体骨架(HPF),然后通过氧化、磷化两步工艺合成了金属原子掺杂CoP的空心多面体框架M-CoP HPFs(M=Ni,Fe,Mn).相较于纯CoP,金属原子的掺杂都可以提高其催化活性,特别是Ni-CoP HPFs的η10(10 mA/cm2处的过电位)只需要92 mV(碱性介质)和144 mV(酸性介质),在10 mA/cm2的电流密度下可以稳定工作21 h.为了揭示金属掺杂对CoP的HER活性的内在影响,基于密度泛函理论计算了掺杂不同金属的(101)晶面的ΔGH*,建立了如图3(A)所示的吸附构型.计算结果表明,CoP晶格中掺入金属杂原子后优化了催化剂对H*吸附的吉布斯自由能,Ni-CoP的ΔGH*趋近于0,因此表现出最佳的催化析氢性能,如图3(B)所示.此外,Wu等[45]在CoP(112)晶面的态密度(DOS)分析表明,当Ni掺杂进入CoP晶格后,d带中心从费米能级下移,降低了H的结合强度,如图3(C)所示.
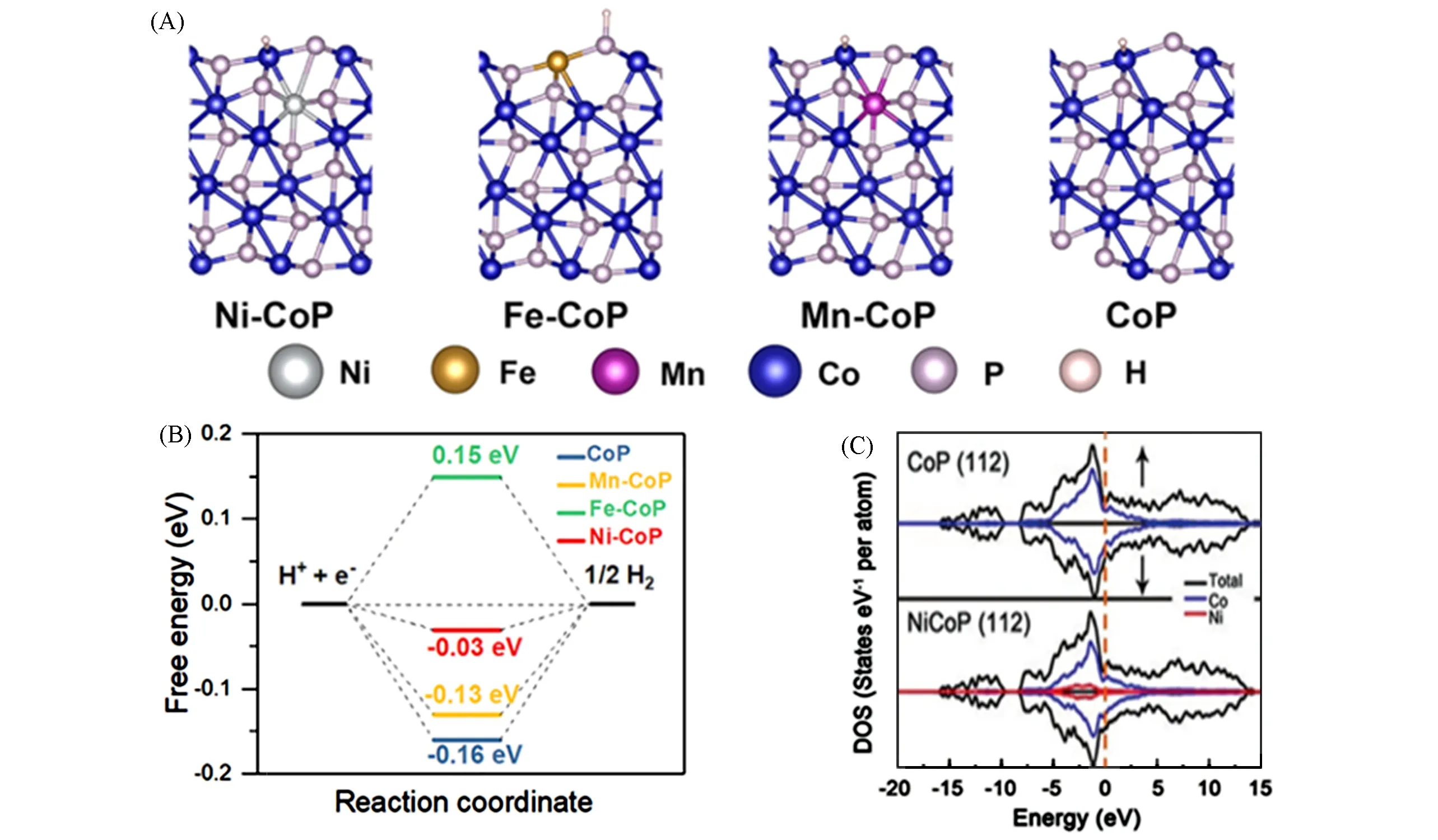
Fig.3 DFT calculationsConfigurations(A)and the calculated free-energy diagram(B)of Ni-CoP,Mn-CoP,Fe-CoP and CoP[44],Copyright 2019,Elsevier;(C)Calculated densities of states(DOS)of Ni0.1Co0.9P and CoP with the Fermi level aligned at 0 eV[45],Copyright 2018,Wiley Online Library.
尽管大多数研究选择本身对具有HER活性的金属掺杂,如第一过渡金属(Mn,Fe,Co,Ni,Cu),但最近的研究表明非活性金属掺杂也具有很好的效果.在密度泛函理论计算的基础上,Sun等[46]提出,在CoP中掺杂Zn可以提高单个活性位点的固有活性.其原因是Zn从相邻的P原子中提取电子,导致电子缺陷位,进而刺激Co原子提供电子密度作为补偿,最终导致Co原子周围的电子密度降低.掺杂后Co原子的DOS计算进一步证明d带中心从费米能级下降,增加了反键轨道的填充,从而降低了ΔGH*.实验结果与理论研究一致,掺Zn的CoP表现出显著的性能提高.同样,将Al引入CoP晶格也可以提高HER的活性.负载在碳布上的Al-CoP催化剂(Al-CoP/CC)在0.5 mol/L H2SO4中驱动10 mA/cm2电流仅需23 mV的过电位,与Pt/C相当,远优于原始CoP/CC(63 mV).在酸性溶液中进行1000次循环伏安(CVs)扫描后,活性损失可以忽略不计.等离子体发射光谱-质谱(ICP-MS)测试表明,在稳定性测试期间,只有12.5%的掺杂Al浸出.Al的部分溶解强调了Al是作为掺杂质存在于CoP晶体结构中,而不是形成极其不稳定的孤立的磷化铝.并推测Al-CoP稳定性的增强可能归因于HER过程中Al的缓慢溶解,增加了表面积,从而补偿稳定性测试过程中的部分HER活性损失[47].
将作为评价电催化反应的描述符d带中心和氢中间体吸附能直接建立强的关联性,可以为催化剂的理性设计提供重要指导.Xiong等[48]在碳布上制备了少量金属(Li,Cr,Mn,Fe,Co,Sn,平均原子比为1.1%)掺杂的M-Ni2P纳米片.DFT计算结果表明,H吸附在Ni空位上是最稳定的构型,也是M-Ni2P表面掺杂金属的近邻位置[图4(A)].电负性越低的掺杂金属对催化剂表面贡献的电子越多,计算得到的ΔGH*数值越低,对H*的吸附能力越强,Bader电荷与ΔGH*具有明显的线性关系[图4(B)].M-Ni2P中Ni原子的d轨道态密度中心变化和电荷转移的方向一致,低电负性杂原子的电子向Ni的费米能级(Ef)附近转移,使Ni原子的d带中心(εd)向Ef移动[图4(C)],H*的s能带和表面金属的d能带之间形成的反键态上移,从而增强对H*的吸附[图4(D)].εd和ΔGH*之间具有明显的线性关系,Co,Fe,Mn掺杂具有适当的ΔGH*,因此可以推断出理想M-Ni2P催化剂的εd应在-2.09 eV左右[图4(E)].在0.5 mol/L H2SO4电解液中测试了不同M-Ni2P催化剂的电催化析氢活性,在电流密度为10 mA/cm2时,样品的过电位如图4(F)所示.低电负性金属掺杂,优化ΔGH*的M-Ni2P催化剂可以提高催化性能,而高电负性金属Sn的掺杂显著降低了Ni2P的活性,催化结果与前面的密度泛函理论计算是一致的.该工作以Ni2P为模型,掺杂电负性不同的金属原子,通过理论计算和实验进一步证明了杂原子造成的电子重新分布可以有效地优化ΔGH*,并证明ΔGH*与d带中心具有良好的线性关系(R2=0.98),通过计算d带中心迁移可以来衡量表面吸附物和过渡金属之间相互作用的强弱变化,从而确定磷化物理想的掺杂金属.
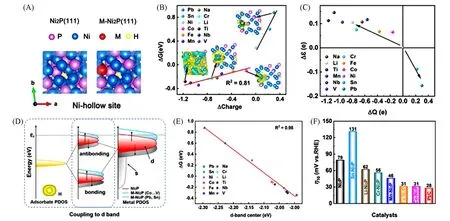
Fig.4 Top view of the H adsorbed on Ni hollow site(A),relationship between hydrogen adsorption free energy(ΔGH*)and charge transfer to the surface(ΔQ)of M-Ni2P[(M=Ti,Nb,V,Li,Cr,Na,Mn,Fe,Co,Sn,and Pb),the insets:charge density difference plots of Pb,Co,Cr and Ti-Ni2P,respectively](B),relationship between εd andΔQ of M-Ni2P(C),corresponding schematic illustration of bond formation of Ni2P and M-Ni2P(D),relationship betweenΔGH*and εd of M-Ni2P(E),and the corresponding overpotentials of Ni2P,M-Ni2P,and Pt/C at a current density of 10 mA/cm2(F)[48]Copyright 2021,Elsevier.
值得注意的是,TMPs中低浓度金属原子掺杂导致的HER性能优化可以通过比较杂原子和主体金属的电负性大小或者未占据d轨道的比例进行预测,然而高浓度金属原子掺杂似乎并不遵循这一规律.Cho等[23]制备了不同过渡金属(Mn,Co,Ni)掺杂FeP的球形纳米颗粒(M-FeP)[图5(A)].通过X射线能谱(EDS)定量元素分析确定每种金属(Co,Ni和Mn)在纳米粒子中的掺杂浓度约为10%,结果证明,仅有掺杂Co的FeP颗粒的HER活性得到提高,反应动力学最快[图5(B)和(C)].Co-FeP性能提升的主要原因可能是Co的引入提高了FeP的电荷转移动力学,Co-FeP的电荷转移电阻(Rct)远小于FeP和Mn/Ni-FeP[图5(D)].因此,不能简单地将杂原子电负性作为提高催化性能的唯一考量,即使是掺杂低电负性的杂原子,也有可能使磷化物主相向更高氧化态迁移[49,50],必须综合考虑杂原子对催化剂整体的影响.
以上的工作主要研究电子分布因素(如电荷转移和d带中心)对H*在催化剂上吸附的调节作用.但H*的来源不同,在酸性介质中质子氢容易获得,而在碱性条件下需要通过水分子在催化剂表面的缓慢吸附解离产生,通常具有较高的势垒[51].所以在碱性电解液中,性能优良的催化剂除了应该具有适当的H*结合强度还应有效地降低水解离的势垒[52,53].设计性能优异的HER催化剂,尤其在碱性体系中,必须同时考虑水解离的因素.
Huang等[54]在泡沫镍(Nickel foam,NF)上制备了具有异质结结构的电催化剂(CoP/CoMoP),经过研究发现CoP中Co位是对于H*,OH*和H2O*的吸附活性位点,而CoMoP和CoMoP-Pv(含P空位)体系中的活性中心是Mo位或Co-Mo空心位.CoMoP和CoMoP-Pv的水解离的势垒降低至0.56和0.57 eV,远低于CoP的水解离势垒(0.84 eV),结果表明,CoMoP和CoMoP-Pv能促进催化剂表面H2O分子解离生成H*,见图6(A).同时,CoP(211)表面适当的ΔGH*也有利于吸附H*的释放,二者形成的界面有利于水的解离和氢气释放,水解离和氢脱附两者之间的协同作用共同促进了催化剂在碱性电解液中的电催化析氢活性.驱动电流密度为10和100 mA/cm2时仅分别需34和94 mV,Tafel斜率仅为33 mV/dec,性能远优于纯CoMoP和纯CoP,电催化结果表明,催化剂水解离和氢吸附能之间的平衡作用有效地促进了碱性电解中的HER反应.Yang等[52]在泡沫镍上制备了具有纳米线阵列的V掺杂CoP析氢催化剂(NF@Co1-xVxP),理论计算表明,在(211)晶面Co1-xVxP需要的H2O解离势垒小于CoP[图6(B)],这表明Co1-xVxP上的H2O吸附和活化的Volmer步骤比在CoP上更容易进行.
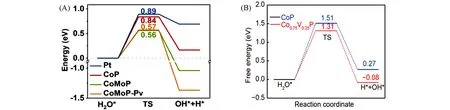
Fig.6 Reaction energy diagram of water dissociation into H*and OH*on the CoP(211),CoMoP(112),CoMoP(112)-Pv and Pt(111)surfaces(A)[54],free energy diagram for H2O activation(cleavage of O—H bonds of H2O molecules)of CoP and Co0.75V0.25P(B)[52](A)Copyright 2020,Elsevier;(B)Copyright 2020,Springer Nature.
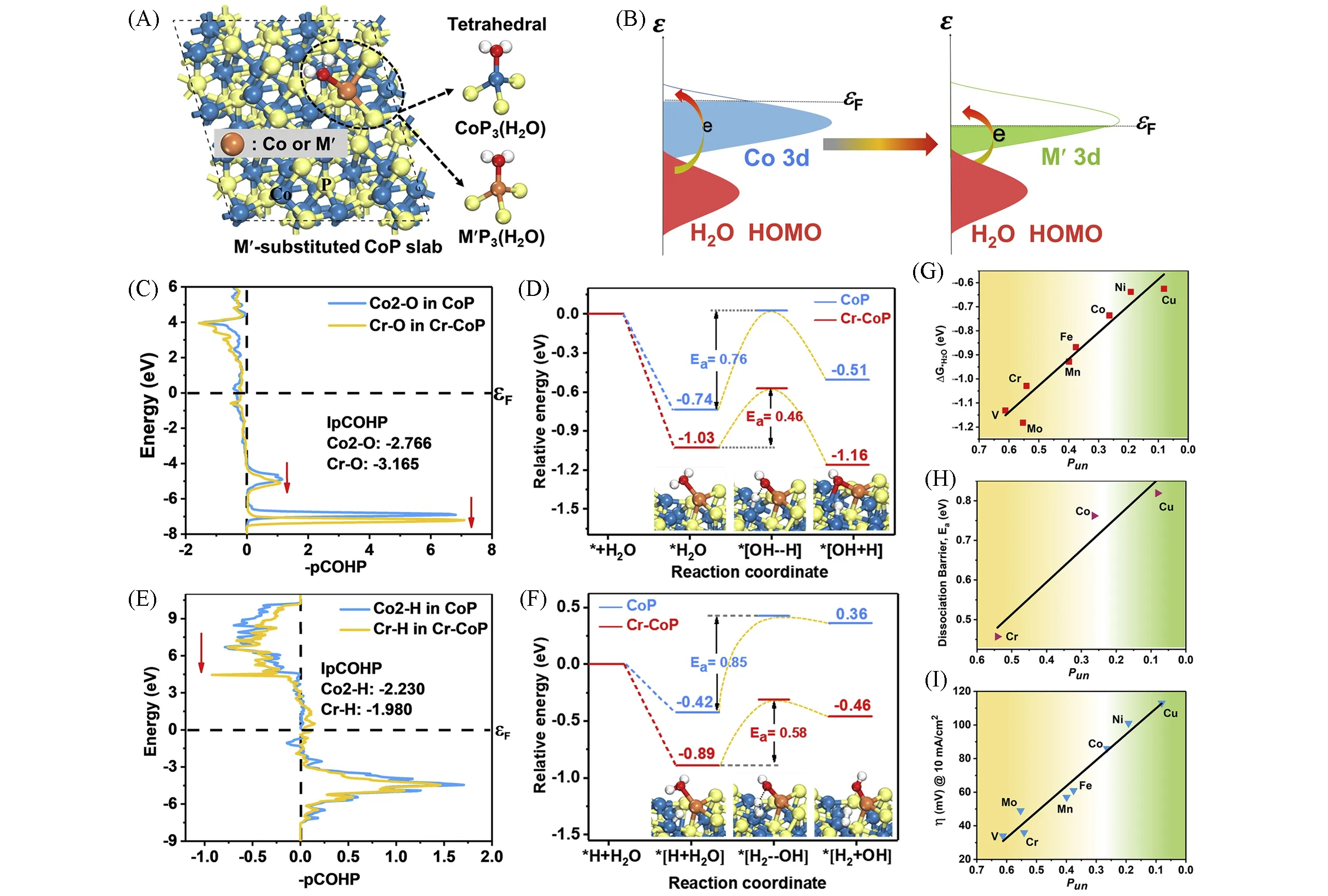
Fig.7 CoP(111)surface model with or without a second transition metal(M′)substitution(A),schematic energy bands of individual water HOMO and M′d orbital on M′-substituted CoP(111)surface(εF:the Fermi level)(B),the crystal orbital Hamilton population(COHP)analysis for the Co2—O bond on CoP(111)surface and the Cr—O bond on the Cr—CoP(111)surface(all of the energies shown relative to the Fermi level εF)(C),the relative energy profiles and the simplified surface structures of the various reaction species along the alkaline Volmer reaction pathway on CoP(111)and Cr—CoP(111)surfaces(D),the COHP analysis for the Co2—H bond in CoP(111)and the Cr—H bond in Cr—CoP(111)(E),the relative energy profiles and the simplified surface structures of the various reaction species along the alkaline Heyrovsky reaction pathway on CoP(111)and Cr—CoP(111)surfaces with an already-adsorbed H(F),the calculated water adsorption free energy(ΔG*H2O)vs.Pun(G),the calculated water dissociation barrier(Ea)of Volmer step vs.Pun(H),trends in η@10 mA/cm2 for alkaline HER shown as a function of Pun(I)[55](A)Blue,orange,and yellow circles represent Co,M′(or Co),and P atoms,respectively;the structures at the upper and lower right are CoP3(H2O)and M′P3(H2O)tetrahedrals,respectively.Copyright 2020,Elsevier.
为了进一步了解金属掺杂剂对碱性介质中HER活性的影响,Luo课题组[55]将CoP作为模型催化剂,系统研究了掺杂多种过渡金属(Fe,Ni,Co,Mn,Cu,Cr,Mo和V)的影响(图7).他们发现活性与掺杂元素的未占d轨道比例(Pun)之间存在很强的相关性.DFT计算表明,原始CoP的Pun较小,这意味着仅存在少量未被占据的Co3d轨道可用于容纳水分子的同能级孤对电子.此外,Co中的高3d电子比例导致其与水的同向电子间存在强电子-电子相互排斥,从而削弱了水的吸附和随后的解离概率.比Co具有更高Pun的掺杂元素的存在可以提供更多未被占据的d轨道,以容纳水的孤对电子,从而加强吸附并启动解离步骤[图7(B)].使用Pun高于Co的Cr进行的计算结果表明,与CoP相比,Cr-CoP的水吸附自由能降低[图7(C)],同时水离解障碍降低[图7(D)].此外,密度泛函理论计算表明,Cr不仅增强了水的吸附/解离,而且还调节了电子结构,其中Cr-CoP比CoP具有更接近热中性的氢吸附自由能[图7(E)和(F)].实验结果与密度泛函理论计算结果十分吻合.掺杂CoP电催化剂在1.0 mol/L KOH中的HER活性增加趋势,与Pun值的趋势完全一致[图7(G)~(I)].该研究强调,过渡金属掺杂元素为主体提供更高的Pun,不仅可以产生亲氧位点以增强水活化,还可以调节主体磷化物的电子结构以赋予优化的H吸附能.
虽然目前对过渡金属磷化物掺杂的研究已经很多,但仍然需要进一步研究掺杂精细结构(如不同位置和掺杂浓度)对析氢活性的影响.为了研究掺杂浓度对析氢活性的影响,Zhang等[56]在碳布上制备了一系列垂直生长的Mo掺杂CoP纳米线阵列(MoxCo1-xP/CC)作为高效HER的催化剂.由于Mo替代Co破坏了本征晶体的结构,纳米线上的晶粒尺寸变小,从而增大了材料的活性比表面积.随着Mo/Co比的增加,MoxCo1-xP/CC的析氢过电位和Tafel斜率产生火山型变化,其中Mo0.25Co0.75P/CC表现出最好的催化活性,且导电性最好.在1 mol/L KOH电解液中,Mo0.25Co0.75P/CC仅需59 mV过电位即可产生10 mA/cm2的电流,并保持稳定的催化活性长达50 h.为了进一步了解Mo/Co比对HER性能的影响,他们选择晶面面积占比最大的(011)晶面解理,计算了MoxCo1-xP(x=0,0.125,0.25,0.375,0.5)的ΔGH*的变化.计算表明,H*中间体倾向于吸附在P原子的顶部位置,如图8(A)~(E),H—P键的结合强度随Mo/Co比的增加而增强,说明Mo掺杂改变了CoP的电子结构,优化了P位置的活性,如图8(G)所示.Mo和Co的原子半径相差很大,当Mo/Co超过一定的比例时,固有的晶格结构受到破坏,由单晶转变为多晶结构,导致电导率下降,抑制了HER活性,因此综合上述所有因素导致Mo0.25Co0.75P/CC具有最好的HER催化活性.
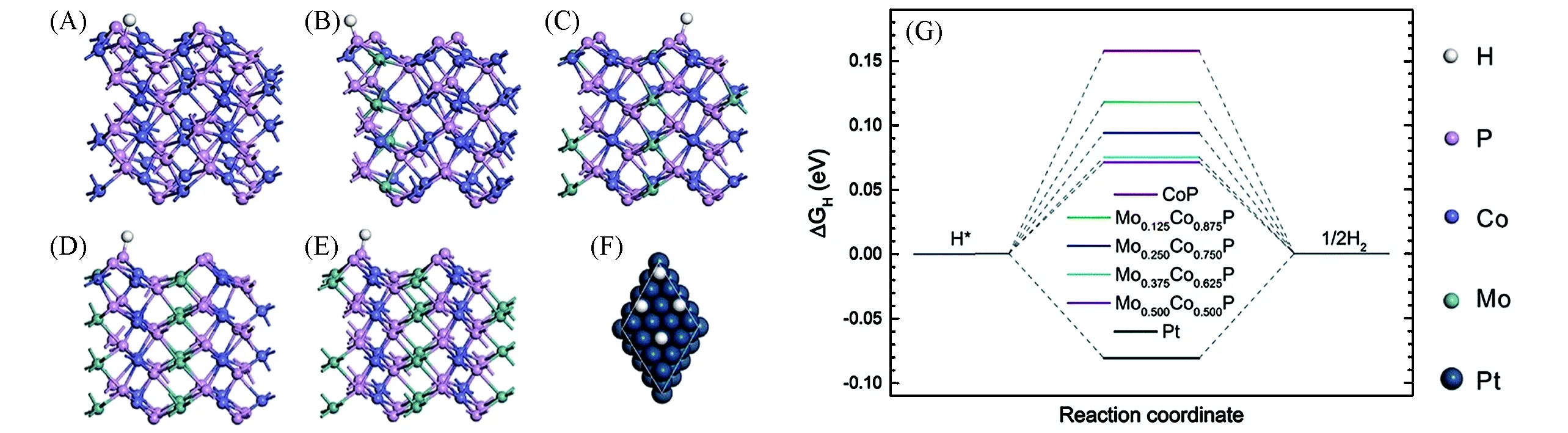
Fig.8 Hydrogen adsorption on the(011)surfaces of CoP(A),Mo0.125Co0.875P(B),Mo0.25Co0.75P(C),Mo0.375Co0.625P(D),Mo0.5Co0.5P(E),and on the surface of Pt(111)(F),free energy diagram of the hydrogen evolution reaction over MoxCo1-xP(011)and Pt(111)(G)[56]Copyright 2019,Royal Society of Chemistry.
Tang等[57]在碳布上制备了不同Fe掺杂CoP的纳米线阵列(FexCo1-xP/CC),计算表明,H优先选择吸附在Co位上,杂原子Fe的电负性略低于Co,削弱了H在Co位上的结合强度,从而使ΔGH*趋于零,当约一半Co被Fe取代时,氢吸附能甚至略优于贵金属Pt,图9(A)~(E).Zhao等[58]通过改变前驱体中Ni/Fe的摩尔比制备了用多孔石墨化碳(GCs)涂覆的双金属磷化物(NixFe1-x)2P@GCs花状催化剂.随着Fe含量的增加,而Ni2p3/2的峰正向移动,元素之间的强相互作用可能会导致局部电偶极子的增加,从而有利于水解物和产物的吸附或解吸.(Ni0.75Fe0.25)2P@GCs具有最快的电荷输运速度,有利于提升HER催化活性.Fe的引入明显降低了Ni2P的氢吸附吉布斯自由能(ΔGH*),而且(Ni0.75Fe0.25)2P的ΔGH*仅为0.181 eV,更接近于热中性,有利于H*的吸附和脱附[图9(F)].同时,(Ni0.75Fe0.25)2P对H2O的吸附能力最强,ΔGH2O为-1.31 eV,有利于促进电解水的Volmer步骤.在1 mol/L KOH电解液中,(Ni0.75Fe0.25)2P@GCs仅需83 mV的过电位即可驱动10 mA/cm2的电流,可以稳定保持催化活性长达30 h.
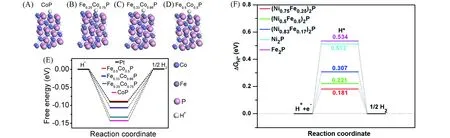
Fig.9 Schematic structural representations for hydrogen adsorption at CoP(A),Fe0.25Co0.75P(B),Fe0.33Co0.66P(C),and Fe0.5Co0.5P(D),free energy diagram of HER under favorable Co site of hydrogen coverage on surface of FexCo1–xP(E)[57],Gibbs free energy image of(NixFe1-x)2P(x=0,0.5,0.75,0.83,1)(F)[58](A)—(E)Copyright 2016,American Chemical Society;(F)Copyright 2019,Royal Society of Chemistry.
此外,Wu等[59]以炭黑粉末作为催化剂载体,在炭黑载体上制备了不同Cr掺杂量的一系列钴基磷化物(CrxCo2-xP/CB).在0.1 mol/L KOH电解液中的HER测试结果表明,Cr0.2Co1.8P/CB仅需218 mV过电位即可产生10 mA/cm2的电流,远低于Co2P/CB 272 mV.DFT计算表明,Cr0.2Co1.8P(201)表面Cr位的水吸附自由能(ΔGH2O*)远低于Co2P(201)面,更容易吸附水分子,表明Cr0.2Co1.8P上的H2O吸附和活化的伏尔默步骤比在Co2P上可能更容易.Cr掺杂造成电子再分布,氢吸附自由能得到优化.掺杂Cr后,Cr0.2Co1.8P(201)总的态密度向右移动,导致费米能级附近的电子密度增加,因此Cr掺杂后的Co2P电子转移动力学更快.得益于掺杂Cr的催化剂对水的吸附活化和对氢吸附优化的协同作用,Cr0.2Co1.8P在碱性溶液中的催化活性明显提升.
另一方面,掺杂可能引起活性中心的改变.在磷化物表面上,H*可以吸附在金属位点、金属桥位、磷位点、金属-磷桥位点或其它类型的位点上,通常通过对比不同位点的ΔGH*,用来确定H倾向于吸附的位点.Meng等[60]利用杂原子Mn调节了CoP催化剂的电子结构,计算结果表明,适当的Mn掺杂量有利于降低催化剂的d带中心.同时通过建立多种可能的掺杂模型,并分别分析了所有活性位点,发现对于未掺杂的CoP(211)表面,H原子倾向于吸附在Co-Co金属桥位点上,但ΔGH*达到-0.15 eV,不利于氢的脱附.当用原子百分比为5%,10%和30%的Mn原子取代Co后,活性吸附位点由桥位转变到了Co的顶点位置,其Co顶点的ΔGH*分别为-0.09,-0.12和-0.14 eV.同时,引入的Mn原子顶点成为新的活性位点,Mn5-,Mn10-和Mn30-doped CoP(211)中Mn顶点ΔGH*分别为0.08,0.05和-0.05 eV,|ΔGH*|值趋向于0,更有利于H的吸附和脱附.因此,引入的杂原子Mn不仅激活了临近的Co原子的活性并且成为新的活性位点,从优化Co位点的氢吸附强度和增加催化剂表面的活性位点数量两方面提高了催化剂的整体催化活性.该研究表明,合适的掺杂浓度和掺杂位置,能够大大增加活性位点的数目,并优化每个位点的本征催化活性.
2.2 双金属掺杂
虽然单一掺杂可以有效地提高电催化活性,但由于其单一的作用,其提高作用仍然非常有限,同时引入两种外来元素可以更有效地发挥调节电子结构和优化表面氢键强度的作用,从而进一步提高催化活性.
Wu等[61]通过管式炉一步加热处理方法将Cr掺杂的FeNi-P纳米颗粒封装在N掺杂的碳纳米管中.在过渡金属的作用下,双氰胺热分解生成的碳被催化转化为碳纳米管,从而起到包裹磷化物纳米颗粒,防止聚集的作用,如图10(A)和(B)所示.Cr-doped FeNi-P/NCN的Fe2p和Ni2p的结合能由于Cr掺杂而正移,改善了催化剂的电子结构,有效地降低了电荷转移电阻.DFT计算表明,掺杂Cr后的催化剂容易吸附水分子,且ΔEH*降低,H*吸附强度减弱,有利于H2的解吸[图10(C)].Cr-doped FeNi-P/NCN仅需190 mV即可提供10 mA/cm2的电流,相对FeNi-P/NCN(334 mV),催化性能提高了43%.

Fig.10 TEM(A),and HRTEM(B)images of the Cr-doped FeNi-P/NCN,adsorption energies of H,H2O,and OH on the FeNi-P and Cr-doped FeNi-P surface for HER(C)[61]Copyright 2019,Wiley Online Library.
Liu等[62]在NF上制备了FeCoP/NF,通过用Ru部分取代Fe位点来调整活性位点之间的相互作用,制备得到Ru-doped FeCoP/NF.由于阳离子之间的电负性差异,Ru掺杂后,Fe原子发生电子转移,出现了较高的铁氧化物状态,如图11(A)和(B)中FeL3-edge X射线吸收近边结构(XANES)谱和X射线光电子能谱(XPS)所示.Ru-doped FeCoP/NF的Rct远低于FeCoP,证明了Ru掺杂有利于提高催化剂的HER动力学过程,电荷容易在电极/溶液界面转移[图11(C)].计算Ru-doped FeCoP/NF的转换频率(TOF)为0.0660 s-1,远高于FeCoP/NF的0.0061 s-1,说明掺杂后催化剂的本征HER活性大大提高,仅需96 mV即可提供100 mA/cm2的电流,催化性能远超商用的20%Pt/C(131mV).
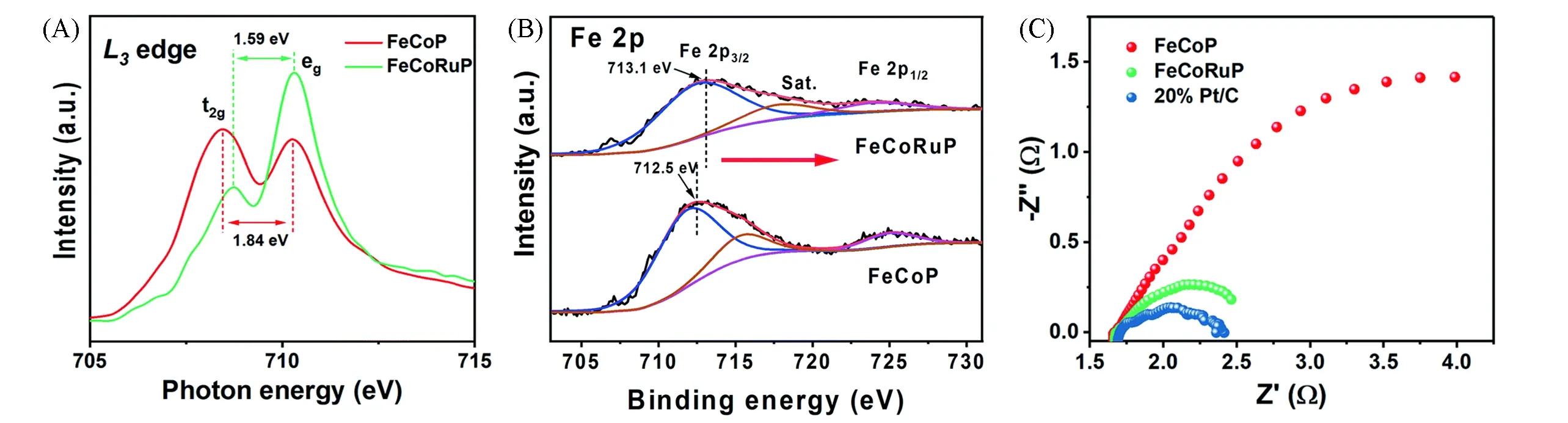
Fig.11 Fe L3-edge XANES(A),Fe2p XPS(B)spectra of FeCoP and FeCoRuP,Nyquist plots of FeCoP,FeCoRuP,and commercial 20%Pt/C(C)[62]Copyright 2020,Royal Society of Chemistry.
双金属掺杂不仅有利于调节催化剂的电子结构,而且有利于增大催化剂的比表面积[63],从而提高电化学性能.Sun等[64]发现相较于CoP NSs,Fe-CoP NSs和Cr-CoP NSs,具有适当双金属掺杂量的Cr0.1Fe0.15Co0.75P NSs具有最好的HER性能,最小的Tafel斜率和最大ECSA,过量的Cr会对Co的电子结构产生影响,过量的Fe会使表面钝化,因此通过实验和理论计算探索适当的杂原子掺杂量具有重要意义.Xiong等[65]研究了V,Cr掺杂量对FeCoP的活性影响,随着杂原子量的增加,催化剂的活性位点密度(Active Site Density,ASD)呈火山型变化,而10 mA/cm2的过电位(η)值在最大ASD处取得最小值[图12(A)和(B)].杂原子不仅可以用于改变磷化物的电子结构以提高内在活性,而且作为牺牲掺杂剂可以暴露更多的活性位点以促进催化反应.Wang等[66]以ZnCo MOF为初始模板,在含Fe2+溶液中进行离子交换,经过退火和磷化后制备得到Fe/Zn-CoP(P/Fe/Co/Zn摩尔比为54.68∶11.17∶22.37∶11.78).HER测试过程中,催化剂上Zn元素在电化学活化过程之后几乎完全流失(P/Fe/Co/Zn摩尔比为33.79∶23.15∶42.54∶0.51),锌的溶解可以暴露更多的活性位点[67],仅需75 mV即可提供10 mA/cm2的电流.
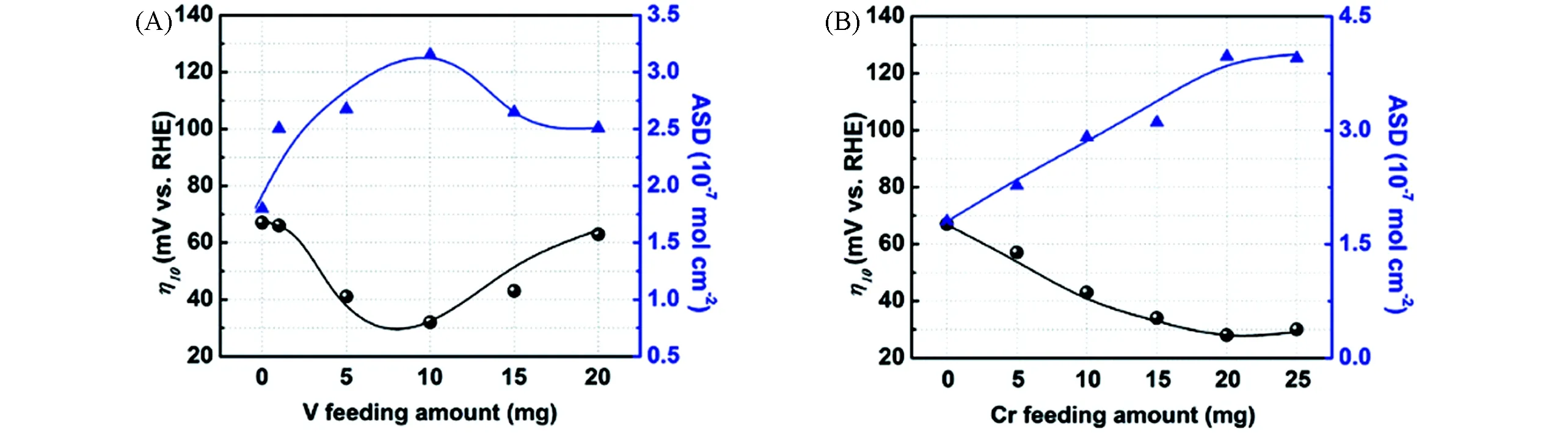
Fig.12 Corresponding overpotentials at a current density of 10 mA/cm2 and the active site density(ASD)with different mol/L doping amounts for V-(A)and Cr-FeCoP(B)[65]Copyright 2020,Royal Society of Chemistry.
2.3 高熵金属磷化物
除了以低浓度杂原子改善磷化物主体金属电子结构的形式制备合金相外,高熵合金磷化物(HEMPs)逐渐引起了科研人员的注意,HEMPs是含有5种均等含量元素的金属组分却只形成一种金属磷化物相的新型电解水催化剂.HEMPs表面原子的几何排列对催化活性产生综合效应(Ensemble effect)[68],合金表面不同聚集体对反应物或中间体具有不同的化学吸附能力,从而为反应过程的不同步骤提供合适的活性中心.
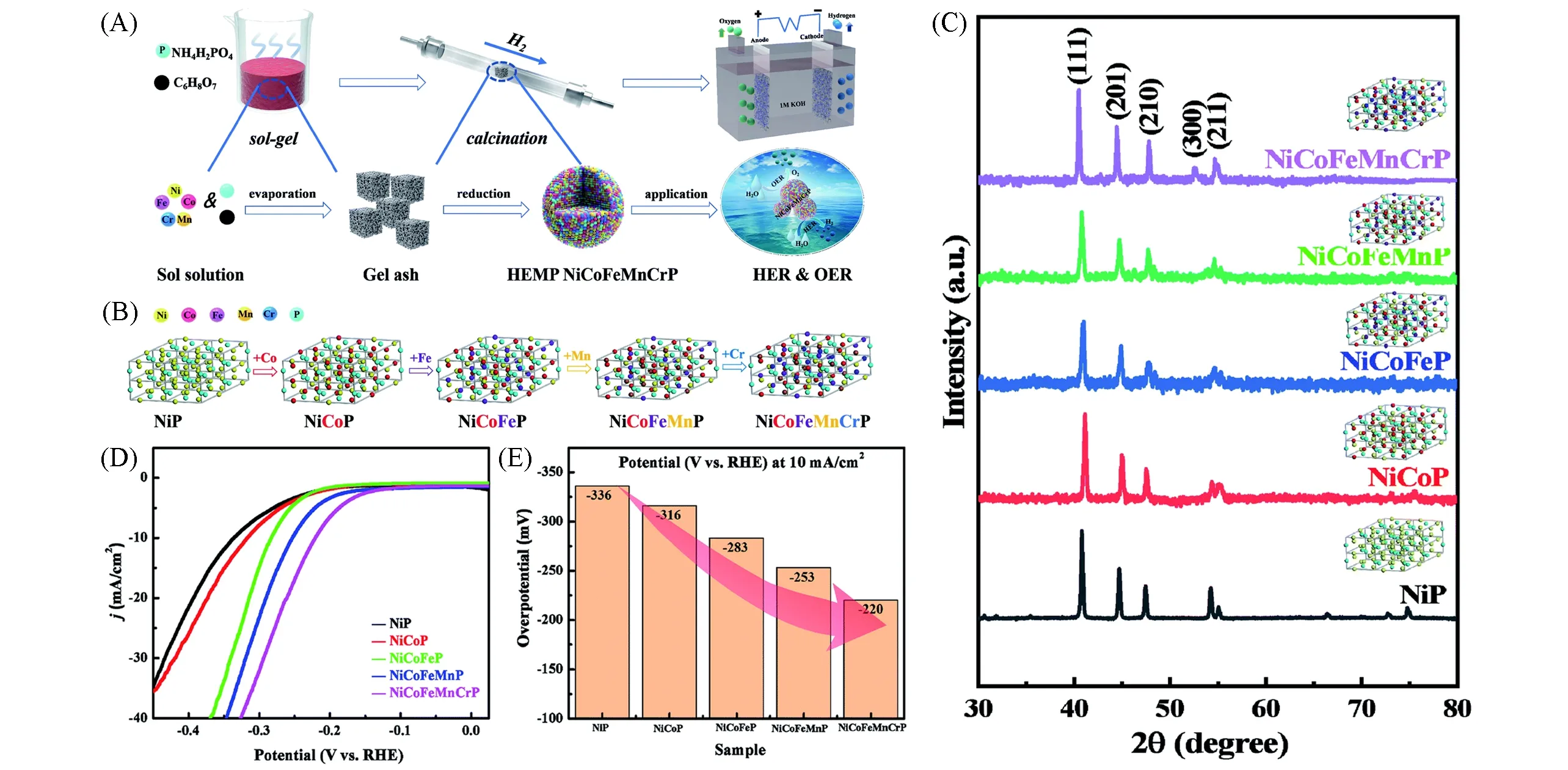
Fig.13 Synthesis process of high-entropy metal phosphide(HEMP)NiCoFeMnCrP NPs for electrocatalytic water splitting through the sol-gel method and calcination reduction strategy(A),the evolution of the crystal structures of NiP,NiCoP,NiCoFeP,NiCoFeMnP and NiCoFeMnCrP NP(B),XRD patterns of NiP,NiCoP,NiCoFeP,NiCoFeMnP and NiCoFeMnCrP NPs(inset:crystal structure model)(C),HER LSV curves(D),and overpotentials at 10 mA/cm2(E)of the different electrocatalysts[70]Copyright 2021,Royal Society of Chemistry.
Zhao等[69]将等摩尔比的Co2+,Cr3+,Fe3+,Mn2+和Ni2+等5种氯化盐和四丁基氯化磷、乙二醇在室温下均匀混合形成共晶溶剂,然后在氮气中于400℃焙烧3 h,最终制备出只有一种单晶结构Fe2P(JCPDS No.51-0943)的高熵合金磷化物HEMPs.10 mA/cm2的电流密度下,HEMP仅需136 mV的过电位,明显优于单金属磷化物Co-P(258 mV),Cr-P(581 mV),Fe-P(497 mV),Mn-P(567 mV)和Ni-P(410 mV),体现出了不同金属之间的协同效应.如图13(A)所示,Lai等[70]采用溶胶-凝胶法将等摩尔比的Co2+,Cr3+,Fe3+,Mn2+和Ni2+等5种氯化盐和磷酸二氢铵(NH4H2PO4)、一水合柠檬酸(C6H8O7.H2O)加水溶解、烘干形成金属原子分布均匀的凝胶灰分,然后在Ar-H2混合气中焙烧得到单一Ni2P相(JCPDS No.74-1385)的高熵金属磷化物NiCoFeMnCrP.作为对比,他们制备了NiP,NiCoP,NiCoFeP和NiCoFeMnP,其XRD结果表明所有的磷化物都是纯的Ni2P相.图13(B)和(C)证明了高熵金属磷化物是金属原子逐步取代Ni原子形成多金属磷化物的过程.如图13(D)和(E)所示,在1.0 mol/L KOH溶液中,NiCoFeMnCrP在10 mA/cm2电流密度的过电位仅为220 mV,远低于中低熵金属磷化物.
从表1可知,掺杂已被证实是磷化物催化剂改性的有效策略.得益于掺杂元素诱导的化学和物理调节,与纯磷化物相比,掺杂后的催化剂显示出增强的活性.因此,各种元素掺杂型析氢电催化剂近几年层出不穷,绝大多数都是金属元素掺杂型电催化剂.因为同周期不同金属阳离子的半径差异较小,同族的性质相似,所以金属元素掺杂相容性通常较好.很多不同金属离子可以相互掺杂,掺杂化合物的稳定性受掺杂比例的影响较小,掺杂相对容易,可以制备出各种金属元素掺杂型电催化剂,甚至多种元素同时掺杂.
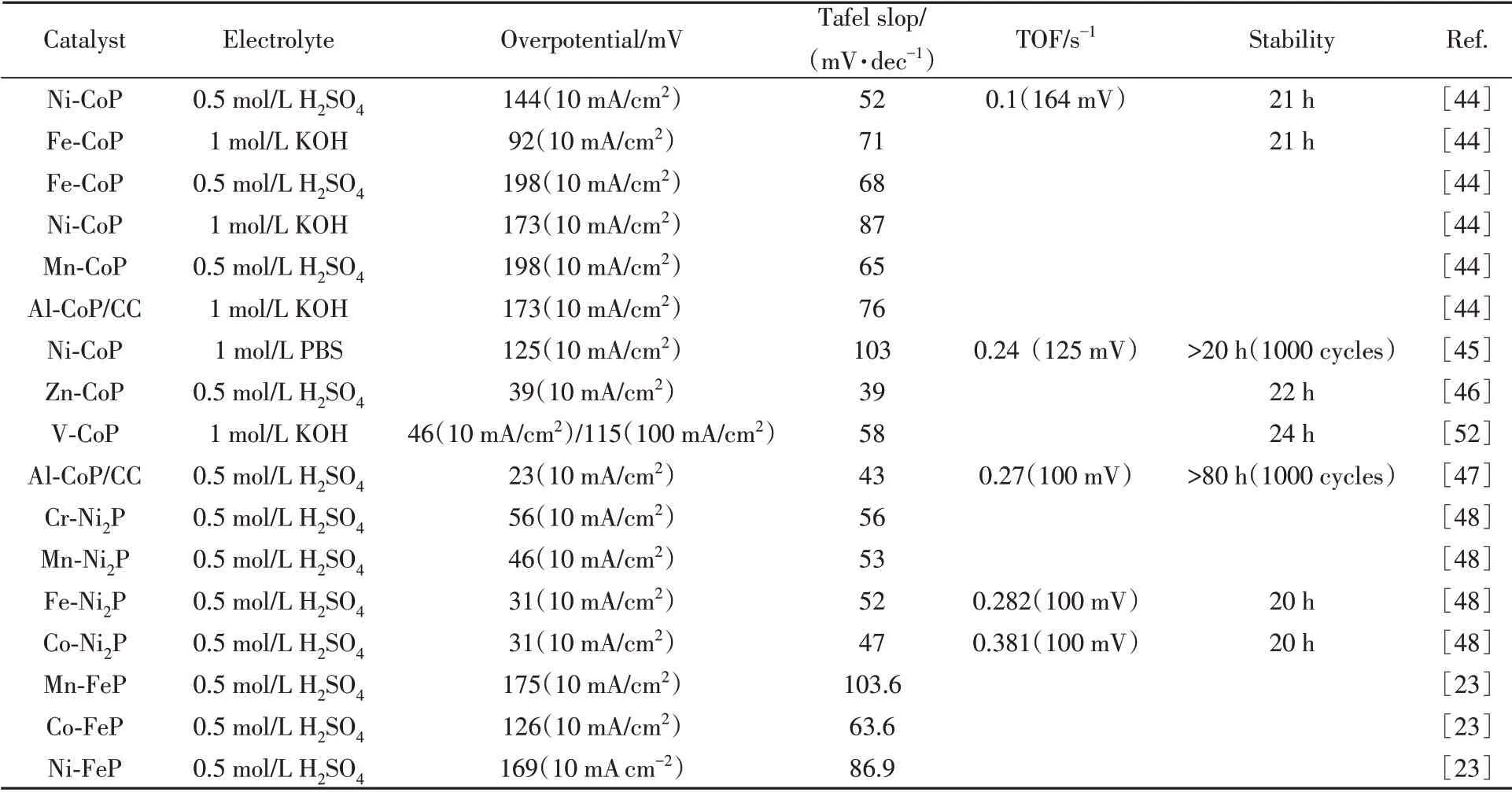
Table 1 HER performance for different metal doped catalysts
3 非金属掺杂
非金属(如S,Se,N,O和B等)具有不同于P的电负性和原子半径,用这些元素取代磷化物主体晶格中的P原子可以引起电子密度在TMPs结构上的重新分布,从而增加电子电导率,并优化反应中间体的吸附自由能,从而提高催化剂的本征活性.此外,合适的掺杂可以增加催化剂的ECSA,提供更多可用的活性中心,进一步提高活性.
3.1 硫掺杂
最常见用于调节电催化性能的非金属掺杂剂为S,可以通过在中温(≤500℃)下利用H2S气体[71]或含S元素的分子化合物[72](如硫脲和硫代乙酰胺)原位分解形成活性含硫物种,煅烧TMP来实现掺杂.Xing等[73]在含有不同量S的碳纸上制造了自支撑S掺杂磷化镍纳米片阵列(S-Ni5P4NPA/CP).S含量对样品的性能有显著影响,掺杂均增加了Ni5P4的ECSA和TOF,硫含量为6%时达到最高活性,仅需要56和104 mV的过电位,便可驱动10和100 mA/cm2的电流密度.XPS分析结果表明,S的引入抑制了HER过程中非活性氧化物的形成,并增加了表面活性中心的比例.此外,由于S和P之间的电子相互作用,使Ni2p向较低的结合能转移.DFT计算进一步支持了电子密度的变化,S-Ni5P4NPA/CP的ΔGH*值变得更接近热中性,促进了反应中间体H*的快速吸附/脱附,从而加速了反应.硫掺杂对稳定性也有重要作用.未掺杂样品在10 h后表现出显著的活性损失,这归因于ICP-MS证实的大量Ni溶解(10 h后为47.1%,100 h后为51.1%).相反,S-Ni5P4NPA/CP在100 h反应后催化性能不降反升(过电位从90 mV降低至70 mV).这种活性增强归因于通过表面重建的逐步自优化过程,S-Ni5P4NPA/CP催化剂100 h后Ni溶出率仅为2.6%,而S则在最低水平上浸出,从而产生空位缺陷和更暴露的活性位点.总之,S掺杂抑制了氧化物的形成和Ni的溶解,并调节了电子结构,有助于提高催化剂的稳定性.
与部分金属元素掺杂类似,S的掺杂也有促进水解离,从而加速电解质中HER的作用.Lee研究团队[74,75]研究了S掺入对CoP[74]和Co2P[75]活性的影响.他们通过XPS分析,证明S分别引起Co2p和P2p结合能的正位移和负位移.DFT计算揭示S掺杂后,费米能级附近的态密度(DOS)降低,这与XPS结果相吻合,表明由于S的掺入使得电子局域化增加,金属性降低.因此,Co中心上的诱导正电荷可以与H2O中O的孤对电子配位,并促进碱性介质HER过程中最缓慢的HO—H键解离步骤.
3.2 氮掺杂
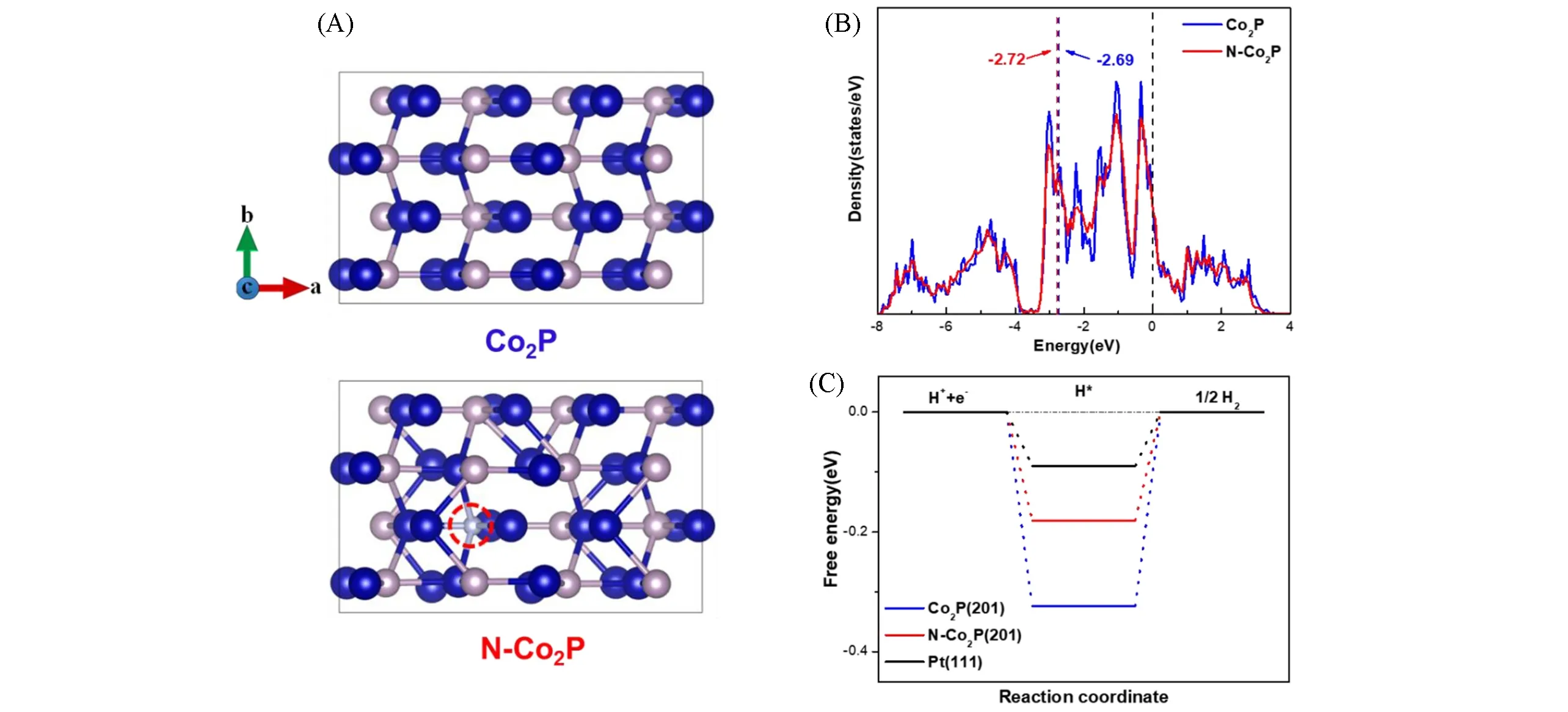
Fig.14 Top views of the optimized geometric structures for Co2P and N—Co2P(Blue ball:Co,pink ball:P,grey ball:N)(A),density of States of Co2P and N—Co2P(B),DFT-calculatedΔGH*for Co2P(201)and N—Co2P(201)surface(C)[79]Copyright 2021,Elsevier.
类似地,可以通过在Ar-NH3气氛下[76]或在尿素[77]存在下加热磷化物材料来合成N掺杂的TMP.通过改变反应参数(如反应温度和时间)以及原料的量来控制掺入的N含量.最近的研究表明,TMP中掺杂一定量的氮均可以改善HER动力学.Luo等[78]在碳布(N-Co2P/CC)上制备了N掺杂Co2P,并在酸性、中性和碱性溶液中进行了测试.优化的N-Co2P/CC催化剂需要27,42和34 mV的低过电位,即可在0.5 mol/L H2SO4,1.0 mol/L PBS和1 mol/L KOH体系下达到10 mA/cm2的电流密度,并且具有较小的塔菲尔斜率和电荷转移电阻,以及长期稳定性.XPS不仅证实了Co—N键的存在,也揭示了N掺杂后导致的有益电子调制:Co2p和P2p信号均向更高的结合能移动了0.4 eV,这表明电子向负电性更大的N中心调节.DFT计算表明,N取代P后,作为水吸附中心Co原子上的正电荷增加,增加了其与水分子中O原子的相互作用.这调节了催化剂对水的吸附自由能,促进了水分子的解离.电子调制还将氢吸附自由能转移到更接近的热中性值,这同样有助于HER的增强.Ji等[79]在磷化过程中加入氮源,对材料进行同时低温氮化,实现N元素的掺杂,并探索了碳酸氢铵的量对掺杂量以及最终性能的影响.XRD,EDS mapping和XPS等结果均表明N元素被均匀地掺入到Co2P中,同时三维多级结构的形貌得以维持,且能形成大量微孔,大大提升材料的电化学活性面积.更重要的是,N元素的掺杂能调节Co2P的电子结构,降低对速控步骤H*吸附的结合能(图14),并减小电子在内部传输的阻力,提高活性位点本征催化活性.在1.0 mol/L KOH中,10 mA/cm2的电流密度处过电位仅为58 mV,Tafel斜率为75 mV/dec.相对于未N掺杂的样品,过电势和塔菲尔斜率分别下降23 mV和7 mV/dec.N-Co2P/NCO/NF具有良好的循环稳定性,5000次循环后,10 mA/cm2处的过电势仅升高3 mV.Zhou等[76]通过DFT计算表明较高的N掺杂浓度会降低CoP的催化活性,即N掺杂存在一个最佳的掺比浓度.
3.3 氧掺杂
近期,氧也被作为有效掺杂元素引入,以提高TMP的电催化性能.氧掺杂的TMP可以通过金属氢氧化物/氧化物的不完全磷化过程合成.如,在不足以使氢氧化物完全磷化的PH3气体存在下加热Co(OH)2纳米片,从而产生含有氧原子掺杂的CoP产物.Tang等[80]通过控制磷化时间制备了O掺杂CoP纳米片(O—CoP),其在1.0 mol/L KOH溶液中10 mA/cm2电流下仅需要98 mV的过电位.此外,与未掺杂材料相比,掺杂催化剂表现出更小的塔菲尔斜率、更低的电荷转移电阻和更高的ECSA,以及优异的稳定性.高分辨率透射电子显微镜(HRTEM)分析表明,CoP内的O产生晶格缺陷(如位错),这是潜在高活性位点的位置.他们通过密度泛函理论计算深入了解了O掺杂对HER活性的作用,DOS计算表明,掺入O后,接近费米能级的电子态增加.这与导电性的增加是一致的,这有助于在O—CoP催化剂上进行HER期间的电荷转移.此外,计算的氢吉布斯自由能变化(ΔGH*)从未掺杂的-0.71 eV,变化到-0.57 eV,反映了掺杂O后更有利的HER过程的中间体吸附/解吸[80].
3.4 硼掺杂
硼的电负性比磷低,得电子能力较弱,能实现了电子密度重新分布,最近发现B的掺杂以前所未有的方式增强了CoP的HER活性.Ren等[81]使用了一种简单的NaBH4辅助还原的方法,在碳纳米管表面进行Co2+还原,其中部分NaBH4原位掺杂在Co前驱体内部,形成B-Co3O4,再经过PH3的冷却磷化过程,成功地将B掺杂在锚定于多壁碳纳米管上的CoP内,最终获得B-CoP/CNT.XPS谱图中存在明显的Co—B键,而没有明显的C—B键信号,表明B掺杂了CoP而不是CNT载体.此外,B掺杂使Co2p3/2和Co2p1/2的结合能负移约0.3 eV.这是由于B从键合Co原子中提取电子的能力比P弱,使得Co氧化状态降低.XAFS结果中,Co K边向较低能量位移,也证实了B-CoP中Co较低的氧化状态.这意味着掺杂后材料中Co原子周围电子密度更高,并间接调制了相邻P原子周围的电子密度,其中P2p出现轻微负位移(约0.15 eV).由于B掺入促进了电子离域,从而提高了其导电性,有利于降低电荷转移电阻,正如Bader电荷分析、电流-电压(I-V)曲线和四点探针测量分析所揭示的,这是催化剂性能提高的关键因素.同时,根据DOS分析,B掺杂后CoP的d带中心下移,CoP(101)晶面的金属Co和非金属P位点的对H吸附强度都得到优化[图15(A)].正如从调制的电子特性所预期的那样,B-CoP在酸性、中性和碱性介质中显示出了显著的性能,分别驱动10 mA/cm2电流,仅需过电位为39,80和69 mV,并具有较高的长期稳定性.
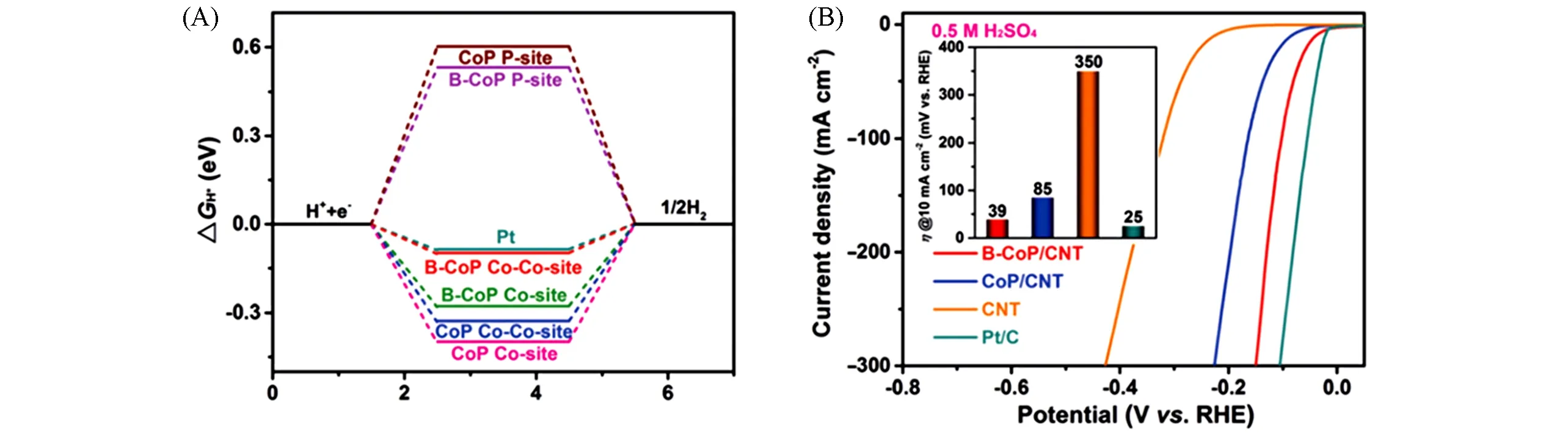
Fig.15 Free-energy diagram for HER(A),and iR-corrected HER polarization curves(B)of B-CoP/CNT,CoP/CNT,phosphatized CNT catalysts and commercialized Pt/C(20%)[81]Inset of(B):the corresponding HER overpotential required for j=10 mA/cm2.Copyright 2020,Wiley Online Library.
3.5 硒掺杂
与S类似,Se也用于改善TMP的性能,然而由于其毒性,相关研究较少.Men等[82]通过计算发现在CoP中引入比P更高电负性的Se阴离子可以降低金属Co中心的3d轨道电子填充度,从而导致Co3d轨道和H2O的HOMO轨道之间产生强耦合,增强的水分子吸附作用有利于H2O活化和解离.在1 mol/L KOH溶液中测试了Se-CoP的HER催化活性,发现在电流密度为10 mA/cm2时过电位仅为41 mV,远低于CoP(91 mV),在200 mV电压下具有0.158 s-1的高TOF值,并具有33 h的超高稳定性.Zhou等[83]通过改变P和Se在热转化反应中与Ni(OH)2反应的量,成功制备了4种具有相同的NiP2立方相结构的催化剂(NiP2,NiP1.93Se0.07,Ni0.09Se1.91和NiSe2),研究了P和Se对活性的协同作用.归因于微量硒引起的电子结构改变,适量掺入Se的NiP1.93Se0.07催化剂表现出最佳的HER活性,具有84 mV的极低过电位(10 mA/cm2)和41 mV/dec的小塔菲尔斜率.
相比金属元素,非金属元素具有更大的电负性,因此掺杂非金属元素,可以使掺杂材料的电子结构和化学性质发生更显著的改变,从而有可能大大提高电催化剂的催化活性(表2).但是,非金属元素掺杂的相容性远不如金属元素掺杂,且非金属元素掺杂容易产生相分离.目前关于非金属掺杂型TMP析氢电催化剂的研究报道相对较少,缺乏系统的研究.
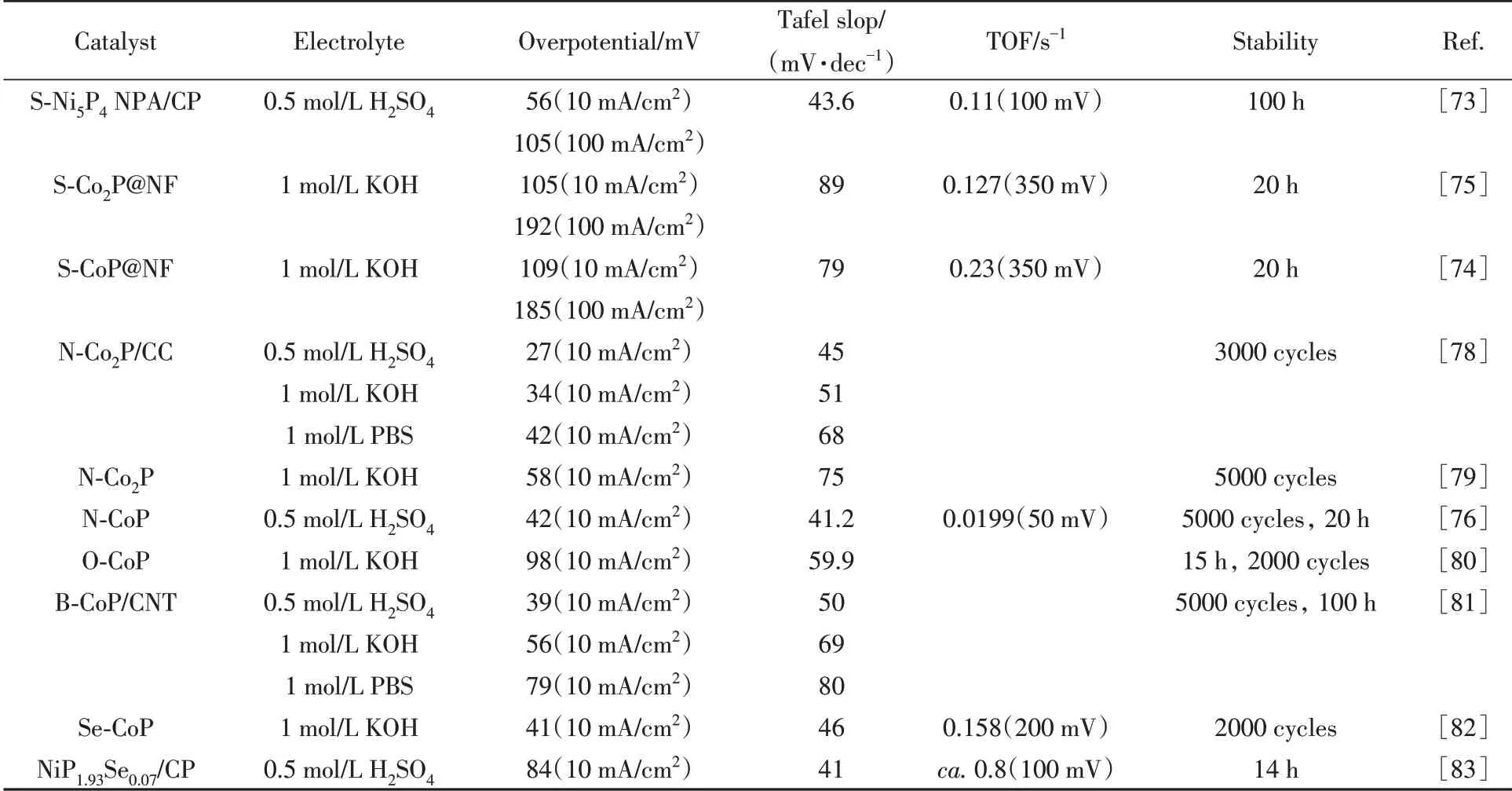
Table 2 HER performance for different non-metal doped catalysts
4 双重掺杂
双重掺杂,即金属和非金属同时掺杂.目前,TMP电催化剂掺杂的大多数研究涉及单一的金属或非金属掺杂剂.然而,最近,人们探索了同时加入金属和非金属掺杂剂调节TMP电催化性能的作用机理.
Cu和O双掺杂策略成功地用于增加CoP的活性位数量,同时增强了水解离和调节H吸附能,已在碱性介质中获得了优异的HER活性.根据XRD分析,Cu和O的存在并没有改变CoP的晶体结构,只是由于晶格参数的降低,导致衍射角稍微向更大角度移动.此外,HRTEM成像显示,与原始CoP相比,掺杂的引入导致晶格畸变,这正是暴露活性位点的区域.在HER的评估过程中,发现Cu含量与活性之间存在明显的相关性,样品中含有5.6%的铜表现出最佳活性(O,Cu-CoP-2).O和Cu的双重掺杂使CoP在1.0 mol/L KOH中具有特别低的过电位(74 mV)和塔菲尔斜率(57.7 mV/dec),与相比CoP(137 mV和76.8 mV/dec),性能显著提高[图16(A)].密度泛函理论计算表明,由于Cu和O的共掺杂,水的脱附能减少,这促进了水分解并加速了反应动力学[图16(B)和(C)],ΔGH*也发生了同样的情况,这意味着后续H2生成的动力学也会加速[84].
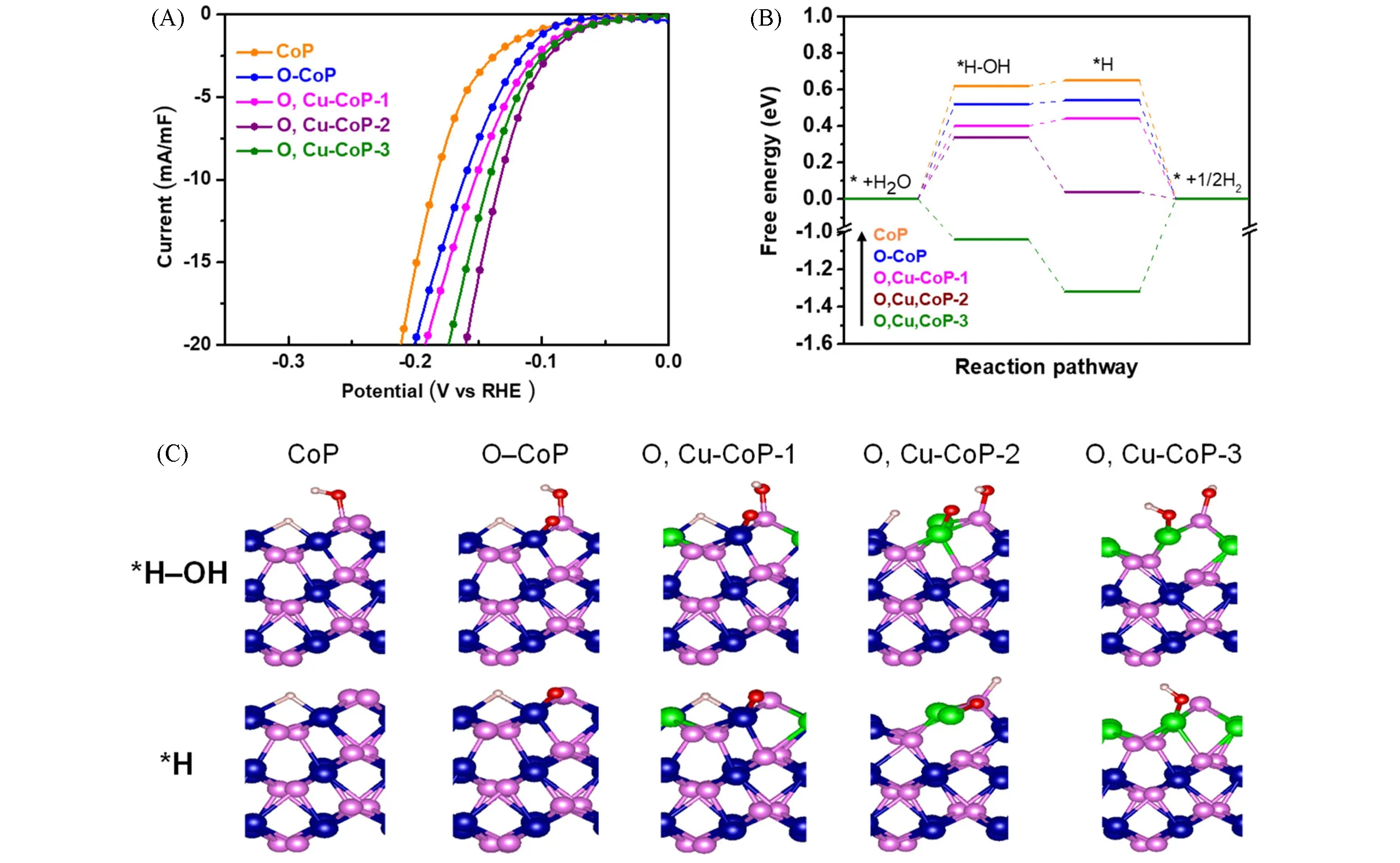
Fig.16 HER polarization curve normalized by electrochemical double-layer capacitance(A),corresponding HER path and energy diagram on the(011)surface of pure CoP and Cu,O-doped CoP(B),structural models of*H-OH and*H adsorption free energies calculated(blue:Co,green:Cu,pink:P,red:O,gray:H)(C)[84]Copyright 2018,American Chemical Society.
Li等[85]制备了支撑在碳纸上的S和Zn共掺杂的CoP纳米棒,并用于整体水分解.优化之后的电催化剂(Zn0.075Co0.925P,S NRCs/CP)在1.0 mol/L PBS体系中驱动HER达到10 mA/cm2电流,仅需要67 mV的过电位,表现优于原始CoP-NRCs/CP(133 mV)和单掺杂催化剂S-CoP-NRCs/CP(106 mV)、Zn0.075Co0.925PNRCs/CP(86 mV).在1.0 mol/L KOH中,S-Zn0.075Co0.925P-NRCs/CP也显示出优异的性能,过电位为37 mV和塔菲尔斜率为41.5 mV/dec.基于材料的结构分析和密度泛函理论计算,解释了S和Zn同时掺杂对活性的影响,优异性能归因于暴露的活性中心数量和电导率的增加,以及对促进H*吸附/解吸过程的电子结构.同样,Lin等[86]也发现在Ni2P中同时掺杂Fe和O可以改善其在碱性条件下的OER/HER性能.
尽管单纯的金属掺杂和非金属掺杂已被证实能有效地提高TMP的电催化活性,但可能是由于电子结构调节不足,这种提高仍受到限制.在这种情况下,预计同时掺入金属和非金属杂原子可能更有效地调节形态和电子结构.此外,由于催化剂中各元素之间的电子相互作用,还能产生协同作用,从而进一步提高催化性能.目前该方面的研究还处于起步阶段,但是预计逐渐会成为研究的热点,并会产生更多更有意义的工作.
5 总结与展望
过渡金属磷化物具有类似氢化酶的性质,是非常具有前景的高效HER电催化剂.通过金属原子或非金属原子掺杂可以有效调节催化剂的电子结构,优化表面的氢吸附强度和改善水的吸附解离.同时也具有降低电荷转移电阻,增加活性比表面积等作用.本文简要总结了掺杂改性的TMP作为HER催化剂的最新进展,探讨了不同种类、数量、位置的掺杂对催化剂的电子结构乃至物理化学性质的影响规律,在催化剂的原子掺杂和催化活性之间建立了桥梁.值得注意的是,尽管在掺杂电催化剂的设计和开发方面取得了重大进展,但未来在多个方面仍然存在挑战:
(1)在材料设计方面应更多地通过计算模拟技术指导催化剂的设计,借助计算机学习高通量筛选可能的方案,并预测催化剂的催化性能.需要综合考虑影响反应的所有可能因素,如,优化氢结合能、促进水离解、优化OH吸附能等,因此必须集成多个计算描述符,将性能和催化剂的结构特性联系起来.理论研究为进一步推进高活性TMP电催化剂的设计提供了有价值的见解.
(2)针对于材料合成,合成具有均匀掺杂剂分布和所需组成的掺杂TMPs催化剂仍然是一个巨大的挑战.精确调制TMP催化剂的结构,包括掺杂剂的类型、数量和位置,需要更加可控的合成策略.此外,除了替代掺杂,其它策略(如簇掺杂或构建杂原子-空位对、周期性的缺陷或掺杂剂的有序分布等)的尝试,显示出越来越大的潜力,值得进一步探索.
(3)对于微观结构和反应机理需要更加深入的理解.在大多数情况下,掺杂调节的研究集中于催化剂物理性质的变化,而较少关注掺杂剂在电催化中的实际功能,引入的掺杂剂是否作为电催化的促进剂或活性位点仍有待评估,相邻掺杂剂间的协同相互作用需要进一步研究.应从结晶态、自旋态、氧化态和掺杂剂的配位环境等更加微观的角度,为深刻理解高性能掺杂TMP电催化剂的本质提供新见解.另一方面,在工作条件下确定催化剂的真实活性位点或实际反应过程也十分重要.包括各种中间体(OH*,H2O*和H*)与催化剂位点的相互作用、HER过程中催化剂表面的结构重构以及离子通过双电层的运动.因此,需要进行更多的研究应用原位操作和表征技术,如X射线吸收光谱或拉曼光谱等手段.
(4)从应用的角度来看,现有的TMP电催化剂仅在人工电解质条件(KOH,H2SO4和PBS)下工作.而在特定地点和特殊环境中,直接海水电解是未来越来越有吸引力的电/氢转换和储存技术.通过杂原子掺入来调节电催化剂以在真实水条件下工作的策略,特别是使用海水作为电解质,具有更加广阔的前景和诱人的吸引力.但是,为了避免掺杂原子在电催化反应过程中优先溶解到电解质中,导致结构破坏和活性消失,TMP尤其是掺杂TMP的结构和活性稳定性在未来应给予更多的关注.