神华煤直接液化工艺中溶剂分子结构解析
2022-11-11范欢欢白佳凯王兴宝李文英
范欢欢,白佳凯,李 旺,王兴宝,冯 杰,李文英
(太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
煤直接液化技术作为石油化工的一种补充技术,对保障我国能源安全具有重大意义[1-4]。煤在高压临氢环境体系,供氢溶剂通过减弱和破坏煤大分子结构网络间/内的相互作用力[5-8],将煤大分子结构溶胀、解离出相对小的分子片段,加速煤氢解反应[9-11],同时,供氢溶剂可将氢原子转移到煤裂解的结构片段,使其进一步发生加氢饱和反应,以期获得合格油品[12-13]。由此可见,煤直接液化技术中供氢溶剂的组成及其结构特点、供氢能力大小,将会直接影响煤液化收率。2008-12-30,煤液化装置以投煤为标志,开启神华煤直接液化一期工程项目的规模化生产油品。供氢溶剂主要由220~350 ℃馏分的中温溶剂和 >350 ℃的高温溶剂混合而得,存在大量同分异构体[14-16]。评价溶剂供氢能力的关键是在反应条件下能否有效解离出自由基氢,这个涉及到溶剂中不饱和结构特性以及不同取代结构中对应的C—H键的解离能[17-21]。鉴于神华煤直接液化工厂的油收率需要进一步提高,笔者结合仪器分析表征结果,通过理论计算分析,从分子水平上揭示供氢溶剂供氢能力大小的根本原因,以期今后为最大程度发挥供氢溶剂能力的溶剂选择提供理论支持。
1 实 验
1.1 材料与分析
(1)原料。选用神华上海研究院中试供氢溶剂(RS-S)和鄂尔多斯工业化供氢溶剂(RS-E)2种供氢溶剂为研究对象。
(2)试剂。正己烷(C6H14,分析纯AR)购于科密欧化学试剂有限公司;二氯甲烷(CH2Cl2,99.8%)购于阿拉丁试剂有限公司。
(3)正己烷可溶物。由于GC×GC-MS/FID的进样口气化温度和色谱柱耐受温度等限制,仅可对沸点320 ℃以下的物质进行检测并获得相对准确的定量分析结果,故对考察溶剂预处理。采用萃取方法对2种供氢溶剂进行索氏抽提,得到正己烷可溶物,再用GC×GC-MS/FID定性定量分析。具体的操作流程如下:将供氢溶剂放入滤筒后置于索氏抽提器中,圆底烧瓶中加入足量的正己烷,组装后置于60 ℃的水浴锅中进行抽提24 h,至抽提器中正己烷呈无色。将圆底烧瓶中的液体进行旋蒸除去正己烷,得到正己烷可溶物。RS-S的正己烷可溶物占89.5%,RS-E的正己烷可溶物占99.1%,因此,分析正己烷可溶物具有一定的代表性。将正己烷可溶物稀释于二氯甲烷中,待用。
1.2 仪器表征与计算方法
1.2.1 元素分析
采用德国Vario MACRO cube元素分析仪的CHNS模式和O模式分别测定溶剂中的C,H,N,S和O元素质量占比,结果见表1。

表1 供氢溶剂元素分析
1.2.2 傅里叶变换红外光谱分析
采用Bruker TENSOR 27型傅里叶变换红外光谱仪测定供氢溶剂分子结构的官能团信息。用液体池对2种供氢溶剂进行测定,分辨率为1 cm-1,扫描范围为4 000~400 cm-1。
1.2.3 同步荧光光谱分析
采用美国Varian公司Cary Eclipse(EL05033883)荧光分光光度计对供氢溶剂的芳香度进行测定,激发光和发射光狭缝宽度均为5 nm,采用固定波长方式进行同步扫描,波长差Δλ=14 nm,扫描范围250~550 nm,速率为600 nm/min,待测样品质量浓度为0.1 mg/mL。
1.2.4 全二维色谱-质谱联用仪分析
采用岛津公司的QP2020型全二维色谱-质谱联用仪(GC×GC-MS/FID)对供氢溶剂进行定性定量检测。色谱柱:第1根,DB-1(15 m×0.25 mm×0.25 μm),第2根,BPX-50(2.75 m×0.1 mm×0.1 μm);进样量为0.6 mL,分流比为30∶1,进样口温度为280 ℃,载气为高纯He,恒定流速为1 mL/min。色谱柱升温程序:初始温度为60 ℃,以3 ℃/min升至300 ℃,保持5 min,调制周期为6 s。MS条件:溶剂延迟8 min。扫描质核比m/z范围:45~400 amu,扫描频率为50 Hz。数据采集软件为Mass Insight,数据分析软件为GC Image 2.3。使用峰面积归一化法定量主要组分。
1.3 密度泛函理论(DFT)计算方法
所有的计算均采用Gaussian 09软件包完成[22],几何结构优化以及频率分析均采用(U)M06-2X泛函进行[23],基组选用6-31G(d,p)。单点能计算在优化后的结构上进行,选用更精确的(U)M06-2X/6-311+G(2df,2p)方法进行计算,并考虑溶剂化效应对能量的影响,溶剂化效应采用SMD溶剂化模型[24]。
焓变计算方程:
H298=E+ZPE+Htrans+Hrot+Hvib+RT
C—H键解离能:
BDE(A-B)= [H298(A·)+H298(B·)] -H298(A-B)
式中,E为电子能;ZPE为零点振动能;Htrans,Hrot,Hvib分别为修正后的热力学参数;R为理想气体常数;T为温度(298.15 K);H298(A·),H298(B·),H298(A-B)分别为自由基A·、B·、中性分子A-B的焓,kJ/mol。
2 结果与讨论
2.1 供氢溶剂分子振动结构分析

2.2 供氢溶剂分子结构同步荧光光谱分析
根据芳香族化合物的特征峰位置[27-28]:270~300 nm为单环芳烃,300~340 nm为双环芳烃,340~400 nm为三环芳烃,400~425 nm为四环芳烃,大于425 nm的为五环或五环以上的芳烃。从图2可见,RS-S的出峰位置主要集中在280~410 nm,说明含有单环、双环及三环芳烃,425 nm之后出现小峰,说明样品中含有少量的五环或五环以上的芳烃。RS-E的出峰位置主要集中在280~350 nm,说明主要含单环和双环芳烃,且340 nm之前出峰明显,说明双环芳烃占比较大,在350 nm后出现小峰,说明样品中存在少量的三环或三环以上的芳烃。
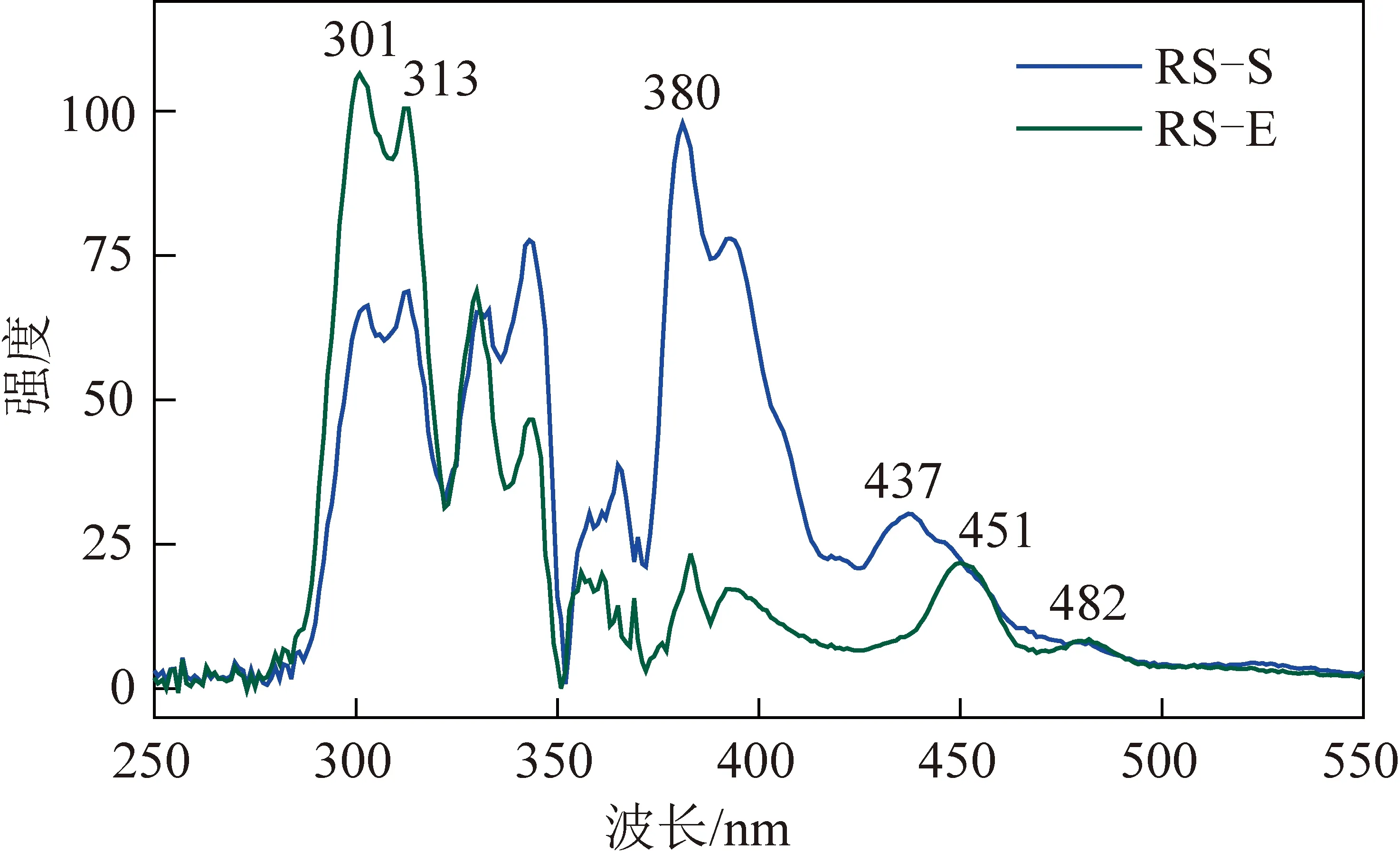
图2 供氢溶剂的同步荧光光谱Fig.2 Synchronous fluorescence spectra of H-donor solvents
2.3 GC×GC-MS/FID定量分析供氢溶剂组成
图3为样品RS-S和RS-E的全二维色谱。对供氢溶剂进行定性分析,发现供氢溶剂主要由链烷烃、芳烃、氢化芳烃、环烷烃等组成。从图3可以看出,RS-S中所含化合物的种类多于RS-E。随着一维方向其碳数从低到高的规律进行排列,二维以极性从低到高进行排列,整体上呈瓦片状分布,最下边为链烷烃分布,碳数依次增加,同时,RS-S的链烷烃种类要多于RS-E。从下往上为链烷烃、环烷烃、芳烃等,从左到右可大致分为单环苯类、双环萘类、联苯类、三环蒽菲类、四环芘类等,从图3(a)可知,RS-S主要含有萘类、联苯、蒽、菲及芘类化合物,从图3(b)可以看出RS-E主要含有联苯、蒽、菲及芘类化合物(其中,1tR,2tR分别为一维保留时间,二维保留时间)。

图3 供氢溶剂的全二维色谱图Fig.3 Two-dimensional contour plot of H-donor solvents
部分氢化的芳烃具有供氢作用,故将样品按氢化芳烃、芳香烃、环烷烃、链烷烃和其他分类,并采用面积归一化法对2种供氢溶剂进行定量分析(图4)。对比发现,RS-S的氢化芳烃含量比RS-E高15.79%,RS-S的环烷烃含量比RS-E低17.11%,说明RS-E加氢过度,造成有效供氢组分环烷基芳烃含量降低,进而影响溶剂的供氢能力,说明RS-S的供氢能力优于RS-E。这也进一步解释了神华上海研究院中试装置的油收率要高于鄂尔多斯煤直接液化工厂的油收率。2种供氢溶剂的杂原子含量均较低,检测出的含杂原子芳香族化合物含量较少。供氢溶剂RS-S和RS-E的链烷烃占比分别为10.32%和13.89%。

图4 供氢溶剂RS-E和RS-S的组成及其相对含量Fig.4 Content of each component of direct coal liquefaction solvents RS-E and RS-S
表2为2种供氢溶剂中主要化合物的含量和结构式,由表2可知,RS-S的主要成分是萘、蒽、菲、芴、芘和联苯及其相应的氢化物,四氢萘系列化合物所占比例最大。RS-E中氢化芳烃的含量明显低于RS-S,饱和芳烃和链烷烃的含量明显多于RS-S。
2.4 溶剂供氢能力与分子结构的关系
2.4.1 取代基位置对四氢萘C—H键能的影响因素
由图1,4和表2发现RS-S中氢化芳烃含量占58.57%,同时供氢溶剂中含有23.74%的烷基类型的取代基,包括2,7-二甲基四氢萘、2-甲基四氢萘等,甲基取代占比8.80%。在液化反应中,C1位上的H最先供出,决定溶剂的供氢能力,为此,分析了—CH3取代四氢萘不同位置对C1—H的解离能(BDEC1—H)及H原子解离后C1上的自旋密度(ρC1)的影响。计算结果表明,1-甲基四氢萘的C1位要比四氢萘更易失去H,相应的键解离能(341.6 kJ/mol)比四氢萘(353.5 kJ/mol)低10 kJ/mol以上,说明—CH3取代1位有利于C1—H断裂,当—CH3取代2,7,8位时C1—H的解离能在352.9~355.5 kJ/mol,此时—CH3取代其他位对C1—H的解离几乎无影响。
—CH3取代1位对C1—H的断裂影响明显,甲基为给电子基,存在诱导效应,同时C1—H与苯环形成σ-π键,形成超共轭效应,而四氢萘的σ-π超共轭效应强于1-甲基四氢萘,使得1-甲基四氢萘上的电子效应要强于四氢萘,2种效应最终导致C1上的σ电子偏离原来的轨道,偏向π轨道,使得C1上电子密度降低,进而C1—H键越断裂。故1-甲基四氢萘的BDE低于四氢萘。电子自旋密度ρ反映该原子上的未成对电子数量,一般正值越大,表示该原子上的单电子越多,因而具有使电子配对的趋势越强。当1-甲基四氢萘断裂C1—H后,C1上的自旋密度为0.69,低于四氢萘H原子解离后C1上的自旋密度0.79,表明1-甲基四氢萘的供氢能力要强于四氢萘的供氢能力。

表2 供氢溶剂中主要化合物分子结构式及含量
2.4.2 取代基对四氢萘C—H键键能的影响
选择不同取代基取代四氢萘1位,考察其对四氢萘C1—H键断裂能的影响。选择给电子取代基:—CH(CH3)2,—(CH2)2CH3,—CH2CH3,—CH3;吸电子取代基:—COOH,—COCH3,—COH,—OCH3,—C6H5。图5为不同取代基取代四氢萘C1位时C1—H键的解离能(BDEC1—H)以及H原子解离后C原子上的自旋密度ρC1,当取代基取代四氢萘1位时,对BDEC1—H的影响最大,吸电子取代基的BDEC1—H在321.3~336.5 kJ/mol,低于四氢萘的BDEC1—H,吸电子取代后可以将所需的解离能降低17.0~32.2 kJ/mol,说明吸电子取代对C1—H的断裂有促进作用。ρC1均低于四氢萘的0.79,说明吸电子可以将C1上的电子密度降低,从而有利于C1—H的断裂。给电子取代四氢萘C1位可以将BDEC1—H降低7.8~11.9 kJ/mol,说明吸电子取代对C1—H键断裂的促进作用比给电子取代对C1—H键断裂的促进作用更明显。2类取代基对取代2,3,4位C2—H,C3—H,C4—H的键解离能与四氢萘的计算结果相差较小,说明2类取代基取代1位显著影响C1—H的断裂。

图5 取代基取代四氢萘1位时C1—H的解离能与H原子 解离后C1上的自旋密度的关系Fig.5 Relationship between BDEC1—H at 1 position of tetralin with different substitutions and ρ value of spin density on C1 after loss of H atom
对BDEC1—H与ρC1进行多项式回归,发现是以一元一次线性相关,线性回归方程为y= 218.1+170.5x,R2=0.83,判定系数F为39.48,说明BDEC1—H与ρC1存在显著线性正相关关系。从图5可以看出,随着ρC1越小,BDEC1—H越低,C—H越容易断裂。
2.4.3 供氢溶剂中芳环数和预加氢程度的影响
结合同步荧光光谱分析和GC×GC-MS/FID分析数据,选择2~4环不同芳环数的部分氢化的芳烃,如四氢萘、二氢蒽、二氢菲、二氢芘等模拟供氢溶剂。同时,为获取不同结构差异化合物供氢能力的规律性,用四氢苯并蒽、四氢芘、四氢萘、四氢苯并蒽、二氢蒽、四氢蒽、八氢蒽等为参考溶剂,计算分析了不同结构对C—H键解离能的影响,结果如图6所示。发现随着芳环数的增加,线状排列的芳烃,如四氢萘、四氢蒽、四氢苯并蒽等BDEC1—H依次降低,有利于C1—H的断裂。以角状排列的9,10-二氢菲、4,5-二氢芘为例,9,10-二氢菲的BDEC9—H为356.5 kJ/mol,4,5-二氢芘的BDEC4—H为348.1 kJ/mol,同样也证明了芳环数的增加有利于C—H键的断裂。这主要是因为芳环数增加,体系中的总电子数增加,总电子的振动能降低,π键增大对C1上的电子的吸引能力增强,有利于未成对电子的离域,进而有利于C1—H键的断裂。9,10-二氢菲、4,5-二氢芘键解离能均高于四氢萘、四氢蒽、四氢苯并蒽,这主要与结构排列的方式有关,线状排列的键解离能要低于角状排列的键解离能。
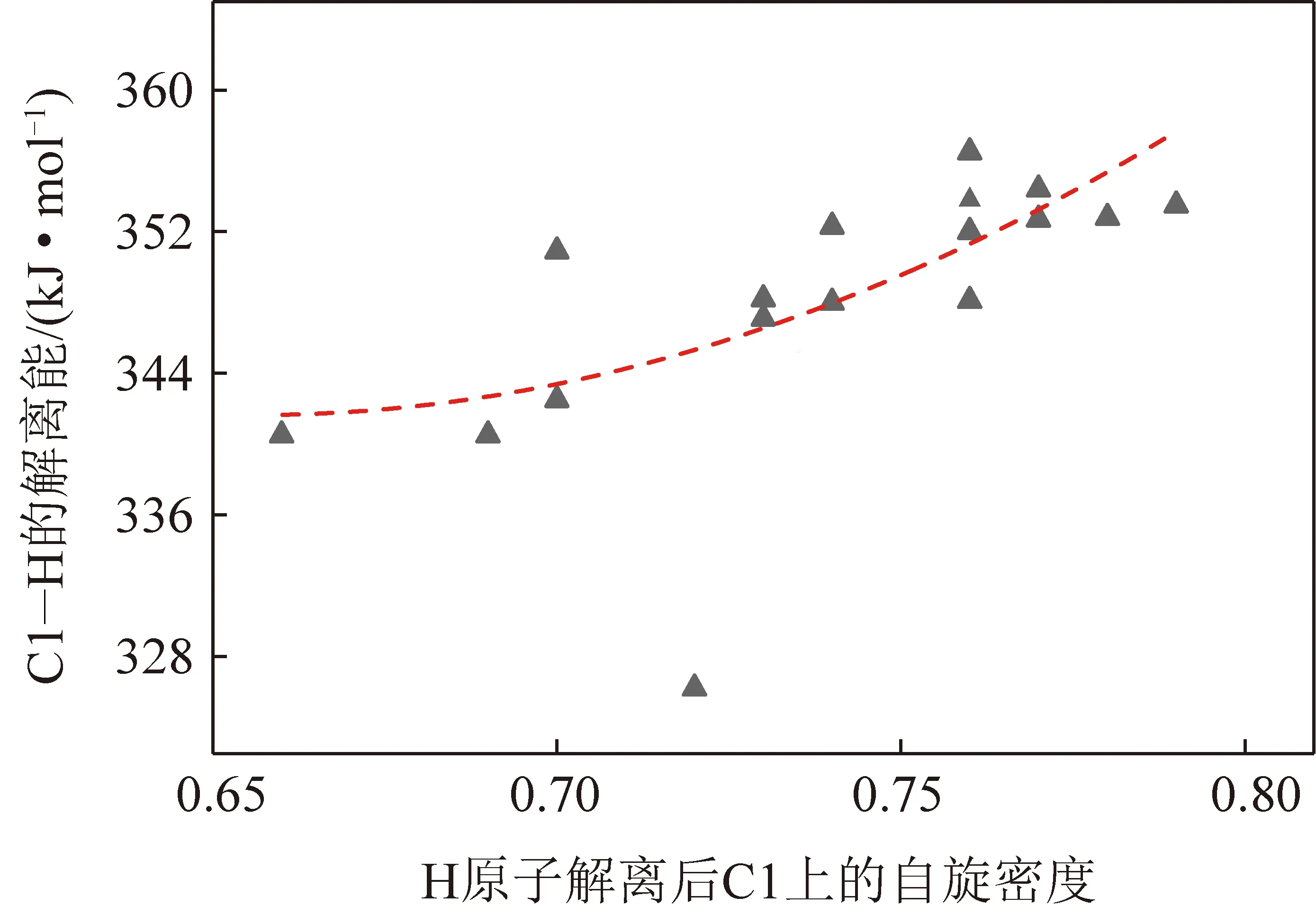
图6 不同结构的氢化芳烃C—H的解离能与H 原子解离后C上的自旋密度ρ的关系Fig.6 BDEC—H of hydrogenated aromatics of different structures and the ρ value of spin density on C after loss of H atom
线性状排列的蒽系列C—H键BDE由大至小顺序为9,10-二氢蒽<1,2,3,4-四氢蒽<1,2,3,4,5,6,7,8-八氢蒽,说明加氢程度越强,对π键的破坏越大,越不利于C—H键断裂。随着角状排列的菲、芘预加氢程度增加,在相似取代位处,键解离能的变化差异小,1,2,3,3a-四氢芘的C3a—H键断裂,键解离能为238.2 kJ/mol,是因为受到周围大π键对C上电子密度的吸引,有利于此处C—H键的断裂。
3 结 论
(1)含有缩合芳环结构以及烷烃结构,芳环数集中在2~4环;有较多不完全饱和的氢化芳烃。
(2)含有碳数小于2的取代基的氢化芳烃;相同化学式氢化芳烃分子,线状排列结构的分子其反应活性要高于角状排列的分子。
(3)由于C—H键键能与解离H原子后C原子上的自旋密度呈正相关性,随着氢化芳环数的增加,溶剂的供氢能力将下降。因此,优良供氢溶剂分子通常含1~3个不饱和芳环。
综上,优良供氢溶剂应是带取代基的、2~4环的、线状排列的不饱和芳烃混合物。
