多取代3,5-二氰基吲哚-吡喃杂环化合物的合成及其晶体结构研究
2022-11-11相艳朱爽石坷鑫李想朱松磊王静
相艳,朱爽,石坷鑫,李想,朱松磊,王静*
(1.江苏省徐州医药高等职业学校,江苏 徐州 221004;2.徐州医科大学,江苏 徐州 221004)
杂环化合物是自然界中分布较广的一类有机化合物,其骨架广泛存在于天然产物和活性化合物结构中,具有重要的药学研究意义[1-3]。文献报道一些含有吡喃环的杂环化合物有许多重要的生理及药理活性,如预防高血压作用[4]、抗过敏作用[5]、抗肿瘤作用[6]等。此外,吲哚类杂环是天然产物中含量最大的杂环化合物之一,具有一系列显著的生理活性[7]。研究表明,多个活性杂环骨架相拼合,往往能表现出更显著的生理药理活性[8]。因此,简便、高效地合成具有潜在生理及药理活性的多样化杂环化合物,对于寻找活性药物先导物的发现和优化有着重要意义[9]。
本论文采用多组分合成策略[10-11],从简单易得的底物出发,利用微波反应技术[12-14],简便快速高效合成2-氨基-3,5-二氰基-4-(对氯苯基)-6-(取代吲哚基)吡喃杂环化合物,通过测定产物的熔点、核磁共振氢谱和高分辨质谱(HRMS)对结构进行了表征。此外,还通过产物单晶的培养和X-射线衍射实验对产物立体结构进行了解析。
1 实验部分
1.1 仪器与试剂
合成反应在Biotage Initiator 微波反应器中进行。熔点(m.p)测定使用XT-5 显微熔点仪;红外光谱(IR)测定使用FT-1000 型红外光谱仪 (Varian公司,用KBr 固体压片);核磁共振氢谱(1HNMR)测定使用日本JEOL 公司400 MHz 核磁共振仪(氘代溶剂采用氘代二甲亚砜,内标试剂为三甲基硅烷);高分辨质谱测定采用Bruker micro-TOF-Q-MS型质谱仪测定;单晶衍射测定在德国Bruker 公司Smart 1000 CCD 仪器上进行。
反应监测用薄层层析(TLC)硅胶板点板,并用紫外灯检测,硅胶板使用青岛黄海开发有限公司,论文中使用的药品和试剂购买于泰坦试剂有限公司。
1.2 合成方法
将2-甲基氰乙酰基吲哚1(1 mmol)、对氯苯甲醛2(1 mmol)和丙二腈3(1 mmol)依次加入到10 mL 微波反应管中,再加入 2 mL 醋酸作为溶剂,预搅拌5 min 后,设置反应温度120 °C,微波加热反应10 min,反应过程采用TLC 法检测。待反应完全后,冷却,取出反应管,将反应混合物倒入饱和食盐水中充分搅拌,静置3 h,析出的固体采用减压抽滤,用少量水和乙醇混合溶剂洗涤滤饼。粗产品在乙醇和N,N-二甲基甲酰胺的混合溶剂中常温下重结晶,得到无色菱形晶体4。
2 结果与讨论
2.1 化合物4 的光谱数据表征
2-氨基-4-(4-氯苯基)-6-(2-甲基-1H-3-吲哚基)-4H-吡喃-3,5-二腈的光谱数据:熔点198~200 °C;IR (KBr):v3 367,3 170,2 192,1 654,1 458,1 398,1 124,754,680;1H NMR (DMSO-d6,400 MHz)δ:2.42 (s,3H,CH3),4.55 (s,1H,CH),7.10 (t,J=7.2 Hz,1H,ArH),7.15 (t,J=7.2 Hz,1H,ArH),7.27 (s,2H,NH2),7.38 (d,J=7.6 Hz,1H,ArH),7.43 (d,J=8.4 Hz,2H,ArH),7.46 (d,J=7.6 Hz,1H,ArH),7.52 (d,J=8.4 Hz,2H,ArH),11.77 (s,1H,NH);HRMS (ESI):calcd for C22H14ClN4O: 385.085 6 [M-H]+,found:385.083 9。
目标产物4 的结构可以从光谱分析上得到表征:红外(IR)谱图中,在3 367 cm-1和 3 170 cm-1处有氨基和吲哚环 N-H 的伸缩振动吸收峰,2 192 cm-1处的强峰为吡喃环上氰基的伸缩振动吸收峰。氢核磁共振谱(1H NMR)图表明,在化学位移值δ11.77×10-6处出现单峰,积分数目一个氢为吲哚环上氮氢峰;在δ7.27×10-6处出现宽的单峰,数目为2 个氢,是氨基吸收峰;在δ4.55×10-6处的单峰,数目为1 个氢,为吡喃环上4 位上的一个次甲基氢吸收峰;在δ2.42×10-6处的单峰,积分数目为3 个氢,是吲哚环上取代基2 位甲基的吸收峰;其余δ(7.10~7.52)×10-6为芳氢,数目与目标产物结构中芳氢数目相吻合。高分辨质谱测得分子离子吸收峰结果和理论计算数值相符合。
2.2 目标产物4 的合成条件优化
该合成是在微波反应条件下,采用三组分一锅法方法进行的,反应10 min 即可完成。同传统热反应相比,微波反应表现出明显的促进作用。同时考察了溶剂和温度等条件对反应的影响。选取乙醇、乙腈、醋酸、DMF、乙二醇和水6 种溶剂进行考察,从表1中可以看出,以醋酸作溶剂中反应产率最高,说明极性的质子溶剂有利于该微波反应。

表1 产物4 的合成溶剂和温度条件优化
此外,还研究了相同醋酸溶剂条件下,不同反应温度的影响:从80 °C 到120 °C,随着反应温度升高,产物4 的产率从60%~88%显著增加,120 °C时反应产率最高,温度继续升高到140 °C 时,产率没有明显增加,保持稳定。因此,该反应的最佳反应条件为温度在120 °C、醋酸做溶剂。
2.3 产物4 的单晶结构解析
通过产物4 的单晶培养,以及X 射线单晶衍射实验进一步确定了化合物的立体结构,为进一步构效关系的研究提供了基础。该化合物主要晶体结构数据列于表2。
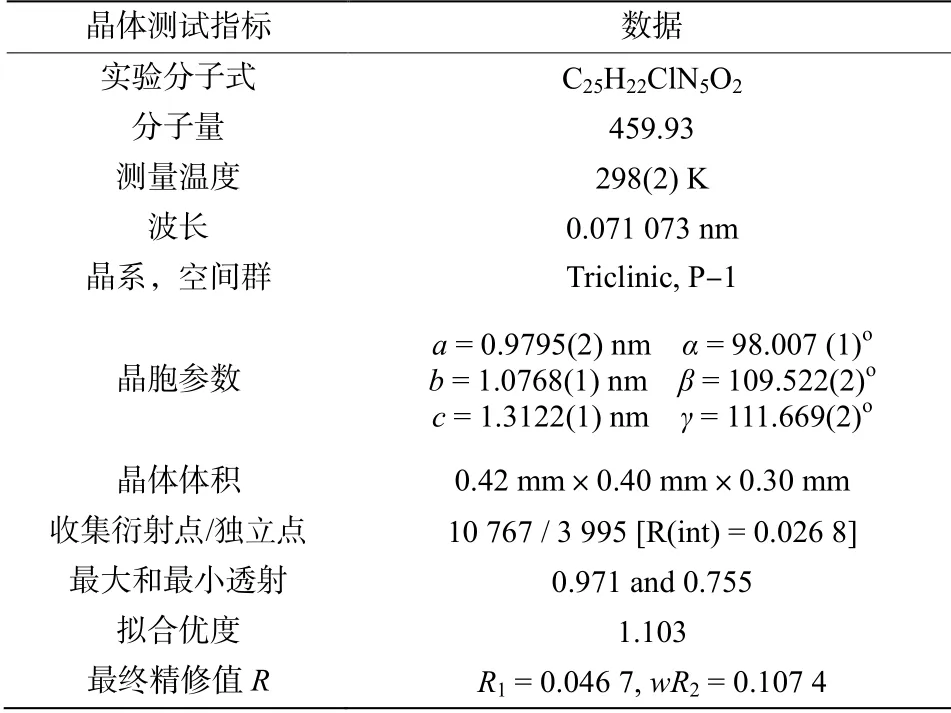
表2 化合物4 的晶体结构数据
化合物4 单晶衍射结果表明:该晶体属于三斜晶系,晶胞空间群为P-1。每个晶胞中含有吲哚环(C(2)-C(5)、N(1))、吡喃环(C(10)-C(14)、O(1))和苯环(C(17)-C(22))等三个环结构,其中吲哚环和苯环原本应为平面构型。吡喃环为反应新构建的杂环,形成吡喃环的6 个原子也几乎在同一平面上,其中最大偏移原子C13 和环平面的偏移距离仅为0.046 6(1)nm。4-氯苯环与吡喃环之间通过C(7)-C(12)原子相连,他们之间的二面角为88.272(9)°接近垂直构型。而2-甲基吲哚环与吡喃环之间以C(3)-C(10)单键相连,两环之间由于吡喃环上5 位氰基和吲哚环2 位甲基的空间位阻作用,使得二者不在同一平面,他们之间二面角为43.731(7)°。此外由于分子间存在着的N1-H1…O2 和N2-H2A…N4,以及N2-H2B…O2 等氢键,晶体的晶胞结构可以保持稳定和延展。图1为产物4 的晶体结构图,图2为产物4 的晶胞堆积图。
3 结 论
杂环衍生物多具有重要的生物药理活性,本论文采用多组分串联反应策略,以微波技术为合成手段,通过简单易得的底物合成了多取代的2-氨基-3,5 二氰基-4-氯苯基-6-(2-甲基吲哚-3-基)吡喃杂环衍生物,产物结构通过了波谱数据表征以及单晶衍射分析。该方法具有反应时间短,操作过程简便,后处理简单且产率高等优点。所得到的产物结构中同时含有官能化的吲哚环和吡喃环等活性杂环骨架,是潜在的药物活性结构单元,其活性筛选和构效关系正在进一步研究中。
