基于PacBio三代测序的香瓜茄(人参果)基因组的组装
2022-11-05司诚钟启文杨世鹏
司诚 钟启文 杨世鹏
(1. 青海大学农林科学院 青海省蔬菜遗传与生理重点实验室,西宁 810016;2. 青海大学农牧学院,西宁 810016)
香瓜茄(Solanum muricatum)是原产于南美洲的二倍体(2n=24)茄科作物[1],因其具有抗氧化、抗糖尿病、抗炎和抗肿瘤活性的作用而闻名[2-4]。果实通常呈圆形、椭圆形或细长形,成熟时黄色的果皮上覆盖着紫色条纹,香气浓郁,果肉黄色带有甜味,芳香多汁,富含大量的维生素C[5],具有较大的营养价值、商业价值以及药用价值。
近几年,香瓜茄作为被消费者熟知的水果作物,其大部分研究工作主要集中在抗病机理[6]、营养价值[7]、药用代谢物[8]、茄科近缘物种比较[9-10]等方面。目前,根据测序技术的不断更迭,番茄(Solanum lycopersicum)[11]、 辣 椒(Capsicum annuum)[12-13]、马 铃 薯(Solanum tuberosum)[14]、 茄 子(Solanum melongena)[15]、烟草(Nicotiana tabacum)[16]等物种的基因组已有多个版本的完整基因组,不同番茄的近缘种(醋栗番茄 Solanum pimpinellifolium[17]、潘那利番茄Solanum pennellii[18])基因组也通过测序被解析。与香瓜茄亲缘关系最近的马铃薯,基于组装了杂合二倍体马铃薯RH89-039-16(RH)之后[19],四倍体马铃薯的第一个高质量单倍型组装也被报道[20]。
植物基因组包含重要的遗传信息。基因组序列可用于植物比较基因组学的研究,同时也是研究植物进化的资源。高质量的参考基因组有利于选择改进农艺性状的基因,对研究其分子机理、加速植物育种至关重要。而Hi-C技术是染色体构象捕获技术结合高通量测序衍生的一种技术,主要研究染色体的三维结构[21]。多种作物基因组序列的公布及解析,为植物重要性状(如果肉性状[22]、抗性水平[23-24]等)以及丰富的基因遗传资源的挖掘等提供了有力工具,还可据此推测基因组的进化演变,促进对关键农艺性状候选基因的筛选和分子标记的开发[25],带动CRISPR等技术的发展及其在作物育种中的应用[26-27],已经成为作物育种改良的重要资源和工具。
目前尚未报道香瓜茄基因组,限制了该物种的各项研究。本研究利用PacBio和Hi-C测序技术,获得香瓜茄的基因组序列,解释香瓜茄与近缘茄科作物的进化关系,为丰富茄科作物基因组信息及进化发育历程,同时为香瓜茄相关分子研究奠定坚实的基础。
1 材料与方法
1.1 材料
材料为香瓜茄甜圆形果实类型(sweet-round friut,SRF)栽培种,采集自青海大学农林科学院园艺创新基地(36°38'N,101°55'E,海拔 2 200 m),经茎尖脱毒处理后,将组培苗新鲜叶片用蒸馏水清洗干净后,擦干,-80℃保存,送北京百迈客生物科技有限公司进行测序。
1.2 方法
1.2.1 DNA的提取 采用CTAB法提取香瓜茄植物组织DNA。
1.2.2 基因组大小的预测
1.2.2.1 文库构建与测序 将检测合格的DNA样品通过Covaris超声波破碎仪随机打断成片段,经末端修复、加Ploy A尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。构建好的文库通过Illumina Hiseq进行PE测序。
1.2.2.2 基因组组装 通过对raw read质控得到clean read,采用SOAPdenovo软件进行拼接。SOAPdenovo拼接的基本过程,利用K-mer频数表数据纠错。对于有低频K-mer出现的reads进行纠错,经过纠错之后的数据用于后续的基因组组装。将纠错后的小片段库的reads截断成更小的序列片段,构建de Brujin图[28-29],获得拼接的contigs。将所有文库测序得到的reads比对回拼接的contigs,利用reads之间的连接关系和插入片段大小信息,将contigs组装成scaffolds。
1.2.2.3 基因组大小预估 利用Illumina HiSeq测序得到测序结果,选取Kmer=41组装到Scaffold,通过K-mer分析初步判断样品的基因组大小、杂合情况、重复序列信息等评估基因组大小。
1.2.3 三代基因组文库构建及组装 打断DNA样品后对打断的DNA样品进行损伤修复及末端修复,连接哑铃型接头,进行核酸外切酶消化,使用BluePippin进行目的片段筛选,获得测序文库。
PacBio测序数据通过初级分析评估、过滤低质量的reads、去除接头后得到reads,进一步碱基纠错后得到高准确性的CCS数据,用于基因组组装、组装后评估等信息分析。
1.2.4 Hi-C技术辅助组装 将香瓜茄组培苗活体植株取样后,利用甲醛将样品固定,将细胞内蛋白与DNA、DNA与DNA之间进行交联,利用限制性内切酶将DNA进行酶切,利用末端修复机制引入生物素标记的碱基,将末端修复后的DNA片段进行环化、DNA解交联及纯化后,破碎为300-700 bp的片段,利用链亲和素磁珠捕获含有互作关系的DNA片段进行文库构建。
文库构建完成后,分别使用Qubit(2.0)和Agilent 2100对文库的浓度和插入片段大小进行检测,使用qPCR方法对文库的有效浓度进行准确定量,以保证文库质量。库检合格后,用Illumina平台进行高通量测序,测序读长为PE150。
1.2.5 基因组注释 对组装完的基因组进行基因组注释,包括重复序列、编码基因及功能注释、假基因、非编码RNA注释等。
1.2.5.1 重复序列注释 采用RepeatModeler2(v2.0.1)[30]、LTR_retriever(v2.8)[31]进行从头预测(从头 预 测 软 件 RECON(v1.0.8)[32]和 RepeatScout(v1.0.6)[33]),RepeatClassifier[30]借 助 于 repbase(v19.06)[34]、REXdb(v3.0)[35]和 Dfam(v3.2)[36]3个已知数据库对预测结果进行分类。将上述从头预测结果和已知数据库合并去冗余后得到该物种特定的重复序列数据库,最后使用RepeatMasker(v4.1.0)[37]基于构建好的重复序列数据库对该基因组进行转座子序列(TE)的预测。
1.2.5.2 编码基因预测及评估 使用Augustus(v2.4)和 SNAP[38]进行从头预测,使用 GeMoMa(v1.7)进行基于同源物种的预测。有参的转录本主要使用 Hisat(v2.0.4)和 Stringtie(v1.2.3)获得,并利用GeneMarkS-T(v5.1)进行基因预测。无参转录本主要通过Trinity(v2.11)[39]组装获得,然后使用PASA(v2.0.2)进行基因预测。最后利用EVM(v1.1.1)[40]整合上述3种方法得到的预测结果。
1.2.5.3 非编码RNA和假基因的预测 非编码RNA包括microRNA、rRNA和tRNA等多种已知功能的RNA,针对不同非编码RNA的结构特点,采用了不同的策略来预测。利用tRNAscan-SE(v1.3.1)识别 tRNA,rRNA 预测主要基于 Rfam(v12.0)[41]数据库并采用barrnap(v0.9)进行预测,miRNA通过miRBase数据库鉴定,snoRNA和snRNA基于Rfam(v12.0)数据库并利用Infenal(1.1)进行预测。通过GenBlastA(v1.0.4)比对,在屏蔽完真基因座的基因组上寻找同源的基因序列,然后利用GeneWise(v2.4.1)[42]寻找基因序列中的不成熟的终止密码子及移码突变。
1.2.6 基因功能注释 对预测得到的基因序列进行 NR(ftp://ftp.ncbi.nlm.nih.gov/blast/db)、KEGG(http://www.genome.jp/kegg)、SWISS-PROT(http://ftp.ebi.ac.uk/pub/databases/swissprot)和 Pfam[41]等数据库的注释分析。
2 结果
2.1 基因组预估及初步组装
2.1.1 测序数据量统计 利用Illumina HiSeq测序得到54.26 Gb的raw reads,经质控后获得54.11 Gb clean reads,测序深度31 X。Clean reads Q20=97.31%,Q30=92.75%,均大于90%,测序错误率(0.04%)<0.05%,也在容错范围内,表明测序质量较好。
2.1.2 17-mer分析及基因组大小估计 通过对香瓜茄过滤得到的54.11 Gb的有效数据进行17-mer分析(图1),根据survey分析结果,在主峰值前约1/2处(depth=15)出现一个较为明显的小峰,说明香瓜茄基因组的杂合程度较高。主峰后depth=62处同样也出现一个小峰,并且与主峰成倍数关系,但由于其峰值较低,峰形不明显,应是重复序列所导致,而非同源多倍体。Depth>62之后的拖尾则是由于香瓜茄基因组重复导致。由公式Kmer-number/depth计算得到的基因组大小约为1 252.41 Mb,修正后的基因组大小为1 238.06 Mb,基因组杂合率为0.84%,重复序列比例为65.87%(表1)。

表1 香瓜茄基因组特征Table 1 Pepino genomic characteristics
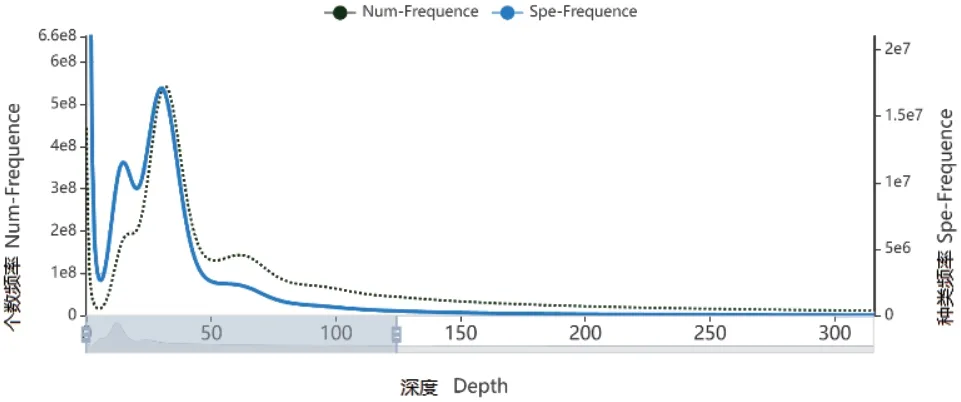
图1 Depth和K-mer个数及种类频率分布图Fig. 1 Depth and number of K-mer as well species frequency distribution
2.1.3 基因组组装结果统计 采用Soapdenovo软件对香瓜茄序列进行拼接(表2),以Kmer=41组装到Scaffold,contig N50为2 049 bp,总长为1 141 353 553 bp,scaffold N50为3 185 bp,总长为1 169 596 440 bp。图2-a及2-b展示contig分布情况。

图2 Contig覆盖深度、长度和数量分布图Fig. 2 Contig coverage depth, length and number distribution map

表2 组装结果统计Table 2 Statistics of assembly results
2.1.4 GC含量及其分布 通过对组装的contig进行GC含量统计,根据contigs的GC分布以及覆盖深度信息绘制散点图(图3)。发现大多分布在20%-50%,主要集中在36%左右,经计算得到基因组GC含量为36.30%。
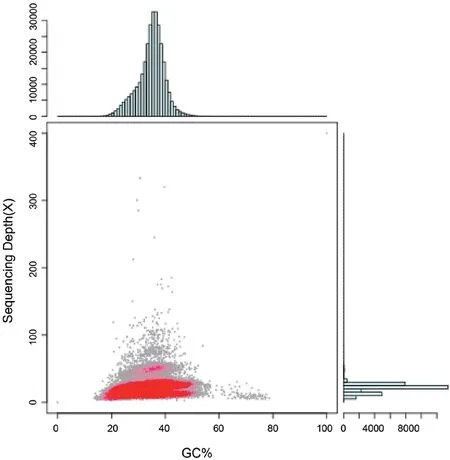
图3 GC含量与测序深度(depth)关联分析统计图Fig. 3 Statistical analysis of GC content and sequencing depth
2.2 PacBio三代测序组装结果及评估
2.2.1 PacBio测序结果 在PacBio测序平台获得香瓜茄基因组的raw reads及组装结果(图4)。使用该样品的基因组DNA构建PacBio文库,测序获得约55 080 918 774 bp(55.08 Gb)的CCS数据,总测序深度约为47.64 X,reads N50为14 640 bp,平均读长为14.179 bp。过滤低质量数据得到的质控后读数共包含3 884 556条reads。

图4 Reads长度分布统计Fig. 4 Reads length distribution statistics
2.2.2 组装结果 PacBio数据进行质控后得到高准确性的CCS数据,然后基于CCS数据使用hifiasm(v0.12)软件进行初步组装,得到基因组序列(表3)。基因组序列总长度为1.15 Gb,contig N50为22.63 Mb,其中,1 kb以上contig数目1 813个,contig N90为596 645 bp,最长的contig为83 851 337 bp,GC含量为35.83%。

表3 组装结果的统计信息Table 3 Statistical information of assembly results
2.2.3 组装结果评估 利用bwa软件将二代高通量测序(如Illumina HiSeq测序平台)得到的短序列与参考基因组比对,统计比对率(99.85%),可评估组装基因组的完整性。使用CEGMA(v2.5)来评估最终基因组组装的完整性,有97.16%的CEGMA基因存在香瓜茄基因组中。使用BUSCO软件评估基因组组装完整性,有98.20%的BUSCO基因存在香瓜茄基因组中,表明基因组组装完整性较高。
2.3 Hi-C辅助基因组组装及评估
对初步组装的基因组序列利用有效Hi-C数据进行进一步的组装,包括初步组装基因组序列的分群、排序和排序后的定向,最终获得染色体水平上基因组序列。共产生143 362 025 128 bp数据。通过Hi-C文库质量评估分析共获得404 890 166对唯一比对到基因组上的reads(unique paired alignments),其中,252 453 038对是有效的Hi-C数据(valid interaction pairs),占唯一比对到基因组数据的62.35%;116 720 788对无效数据中末端悬挂类型的Hi-C数据(dangling end pairs),占唯一比对到基因组数据的28.83%;6 442 178对无效数据中属于相邻连接类型的(re-ligation pairs),占比1.59%;1 535 133对无效数据中属于为自连类型的(self-circle ligation pairs),占比0.83%;27 739 029对无效数据中属于其他未定义的(dumped pairs),占比6.85%。
经过Hi-C组装和人工调整后,共有1 123 245 570 bp的序列长度的基因组序列被定位到12条染色体上,占比97.16%;在定位到染色体上的序列中,能够确定顺序和方向的序列长度为1 079 553 436 bp,占定位染色体序列总长度的96.11%。对Hi-C纠错和组装后得到的基因组序列进行统计(表4),获得最终版本的基因组组装统计结果,Contig N50为22 628 432 bp,Scaffold N50为87 253 278 bp。

表4 香瓜茄Hi-C组装的基因组信息Table 4 Hi-C assembly information of the pepino genome
对于Hi-C组装到染色体的基因组等长切割成500 000 bp一个bin,然后任意2个bin之间覆盖Hi-C Read Pairs的数目作为2个bin之间交互的强度信号(图5),可以明显区分出12个染色体分组;在每一分组内部可以看出位于对角线位置的交互的强度要高于非对角线的位置,说明Hi-C组装的染色体结果中邻近的序列间(对角线位置)交互强度高,而非邻近的序列之间(非对角线位置)的交互信号强度弱,与Hi-C辅助基因组组装的原理一致,证明香瓜茄基因组序列挂载率高。

图5 香瓜茄基因组Hi-C组装染色体交互热图Fig. 5 Hi-C assembly chromosome interaction heat map of pepino genome
2.4 基因预测结果
对组装完的基因组进行基因组注释,包括重复序列、编码基因及功能注释、假基因、非编码RNA注释等。重复序列注释主要包括串联重复序列(tandem repeats)和散在重复序列(interspersed repeats),其中,第二类主要是转座子序列(transposable elements,TE)是研究的主要对象。将从头预测结果和已知数据合并去冗余后得到该物种特定的重复序列数据库,最后基于构建好的重复序列数据库对香瓜茄基因组进行TE的预测。最终得到约742 491 882 bp的TE,占比64.22%,最终得到约201 341 835 bp的串联重复序列,占比17.42%。
对香瓜茄基因采用同源预测、从头预测和转录组预测,基因预测结果(表5)显示,编码基因预测最终得到41 571个基因;非编码RNA即不编码蛋白质的RNA,包括miRNA、rRNA和tRNA等多种已知功能的RNA,针对不同非编码RNA的结构特点,采用了不同的策略来预测不同的非编码RNA,总共预测得到4 360个tRNA、5 677个rRNA、154个miRNA、202个snRNA、287个snoRNA;假基因预测得到449个。利用拟南芥、辣椒、番茄、潘那利番茄以及马铃薯等开展同源预测香瓜茄基因信息,其中,香瓜茄与近缘作物马铃薯预测得到的基因数量最多,有51 586个。

表5 香瓜茄基因预测结果Table 5 Prediction results of pepino gene
BUSCO中embryophyta数据库包含1 614个保守的核心基因。使用BUSCO(v4.0)软件来评估基因预测的完整性,其中,有98.64%的BUSCO基因存在预测的基因中,说明基因预测的完整性高。
2.5 基因功能注释
香瓜茄中99.06%的基因可以注释到所有数据库中(表6)。通过GO注释分析(图6),共有30 713个基因具有GO注释预测的功能,占预测到总基因数的73.88%。GO注释结果显示整个分类中基因分布在细胞组分(cellular component)的相较于分子功能(molecular function)和生物学过程(biological process)较少,生物学过程最多。其中,二级功能分布在细胞内(intracellular)、细胞结构体(cellular anatomical entity)、催化活动(catalytic activity)、结合(binding)、代谢过程(metabolic process)、细胞过程(cellular process)的基因数目相对较多。

图6 GO二级节点注释分类统计图Fig. 6 Statistical chart of GO secondary node annotation classification
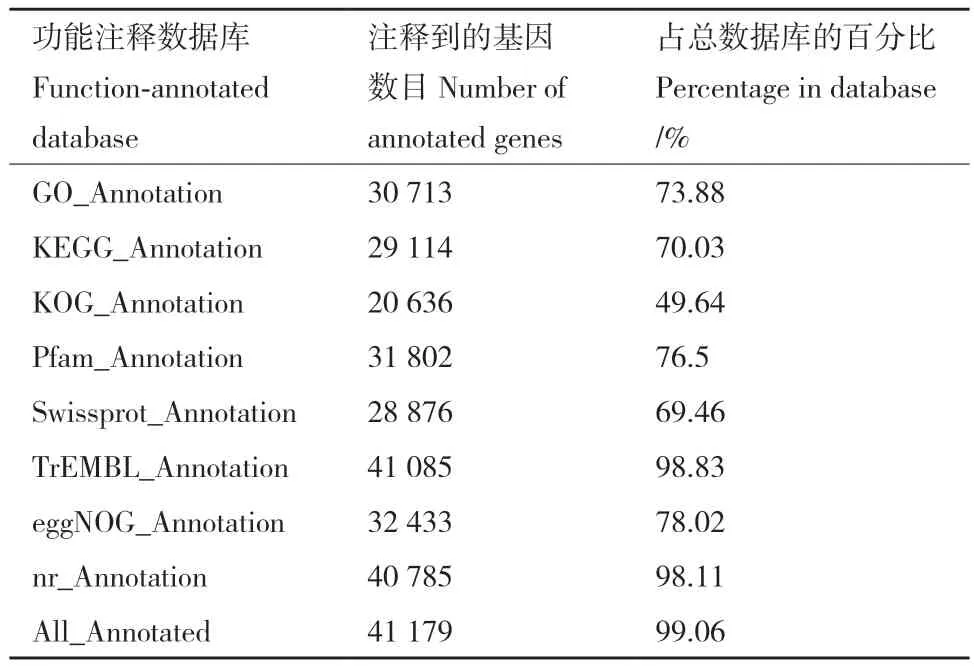
表6 香瓜茄基因功能注释统计信息Table 6 Statistical information of pepino gene function annotation
eggNOG注释结果(图7)显示,香瓜茄的蛋白序列功能主要集中在复制、重组和生物生成(L:replication,recombination and repair),占比 10.94%,转录(K:transcription)占比7.58%,信号转导机制(T:signal transduction mechanisms)占比7.1%,翻译后修饰、蛋白质周转、分子伴侣(O:posttranslational modification,protein turnover,chaperones) 占 比6.82%,能量的产生和转换(C:energy production and conversion)占比5.36%。eggNOG采用了COG,KOG和arCOG中引入的20个功能类别,在功能层面上对基因进行分类。eggNOG结果反映在不同的功能类别中,通过基因数目的多少能够展示出该物种在进化过程中对环境的适应性。
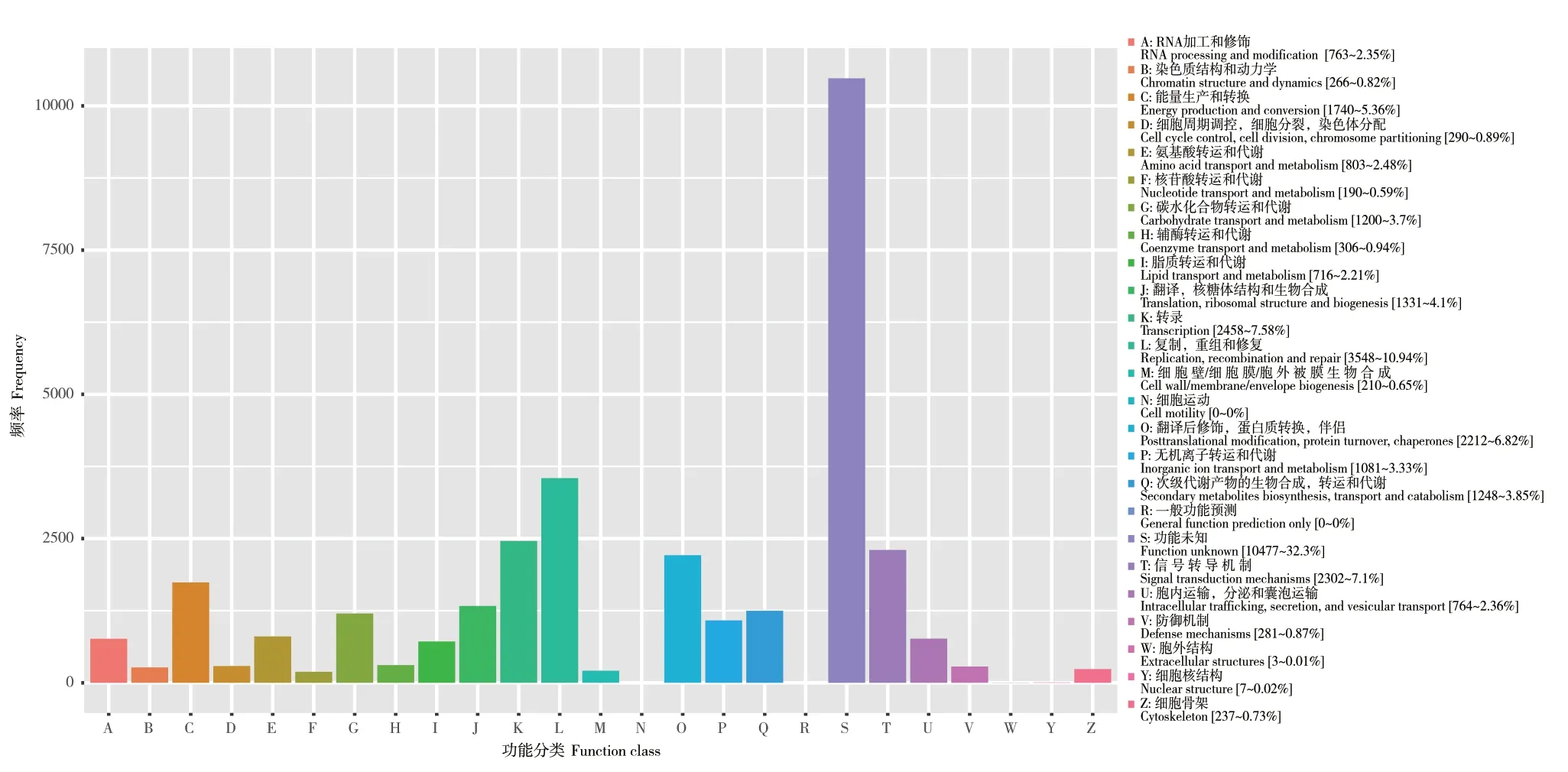
图7 eggNog功能注释分类统计图Fig. 7 eggNog functional annotation classification
2.6 物种进化分析
选择8个已知基因组信息的物种,构建系统进化树。结果表明,香瓜茄与马铃薯的进化时间大约在12.82 MYA(图8)。从进化时间上来看,香瓜茄的进化时间稍晚于烟草、辣椒和茄子。对比于基因预测结果(表5),近缘物种马铃薯作为香瓜茄同源预测物种,预测到的香瓜茄上的基因个数也最多,揭示了马铃薯与香瓜茄较近的进化关系。
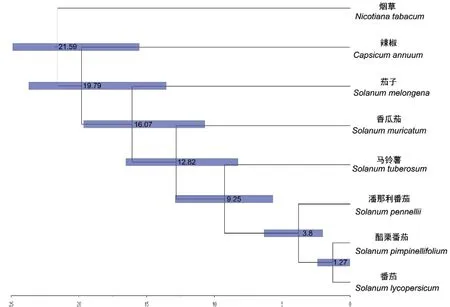
图8 物种间分化时间Fig. 8 Differentiation time between species
3 讨论
连续性和完整性是基因组组装的重要指标,PacBio基因组组装和Illumina数据的纠错可以大大提高测序数据连续性和完整性[43]。本研究通过这种策略对香瓜茄基因组组装显示出高度分辨的结果,N50=22.62 Mb(megabases),与近期测序的芒苞草 N50 = 6.96 Mb[44],黑麦 N50>29 Mb[45]和板蓝根N50=36.16 Mb[46]结果相近。在定位到染色体上的序列中,能够确定顺序和方向的序列长度为1 079 Mb,占定位染色体序列总长度的96.11%。BUSCO和基因预测结果分析进一步证实了香瓜茄基因组的高质量和完整性。
根据基因组数据构建了香瓜茄的系统发育关系,同时对比基因预测结果,香瓜茄比对到马铃薯上的基因个数最多,发现在茄科作物中,香瓜茄与马铃薯进化关系最近,这与先前发表的系统发育分析的结果一致[9]。茄科作为双子叶植物中最重要的果蔬类群,其包含的较多物种的基因组或基因组草图已经被测序完成[47-48]。基因组测序技术及生物信息技术的不断发展[49],显著推动了香瓜茄这种非模式作物的测序研究,香瓜茄基因组的测序完成是对茄科基因组研究的又一补充。我们的研究结果将为茄科的起源,进化和多样化分析增加了功能见解。
本研究通过对香瓜茄基因组的预估,对比PacBio第三代测序技术测序以及Hi-C辅助基因组组装结果,首次揭示了香瓜茄基因组的大小。根据各分析指标,推测香瓜茄基因组为高杂合基因组,针对一些物种基因组重复序列偏多的特征,可以采用三代测序或者HiFi测序等兼顾长读长和高精度的测序手段开展基因组研究。随着长度测序的出现和完善,基因组组装的数量和质量正在不断提升,但一些具有显著生态价值和较低经济价值的植物中参考基因组的数量和质量仍然较低,在137个植物目中,有76个植物目缺乏代表性的参考基因组,62个目至少有1个参考基因组。例如,十字花科目有83个种的参考基因组,禾本目和唇形目分别有80个种和67个种的参考基因组。伴随着技术的进步和越来越多其他物种的关注度提升,未来完整的植物基因组测序数据库构建将成为可能。
4 结论
获得香瓜茄高质量染色体水平参考基因组,推测该测序香瓜茄基因组为高复杂基因组。香瓜茄与马铃薯具有较近的进化关系。
