细菌耐药:生化机制与应对策略
2022-11-05刘成程胡小芳冯友军
刘成程 胡小芳 冯友军
(1. 西安交通大学基础医学院,西安 710061;2. 南昌大学抚州医学院,抚州 344000;3. 浙江大学医学院,杭州 310058)
抗生素作为20世纪最重要的医学发现之一,拯救了无数人的生命,为人类传染病的防治做出了重要贡献。然而抗生素在临床、农业及畜牧业的过度使用和滥用,以及抗生素诱导广谱杀伤的内在能力导致多重耐药(multidrug-resistant,MDR)细菌的出现。这些细菌在世界范围内迅速蔓延,已经成为严重的公共卫生问题。
抗生素过度使用和滥用是导致抗生素耐药(antimicrobial resistance,AMR)的根本原因。鉴于严峻的AMR趋势,2017年世界卫生组织(World Health Organization,WHO)发布了最具耐药性、对人类健康最具威胁的12种“超级细菌”,其中耐碳青霉烯类抗生素的鲍曼不动杆菌、铜绿假单胞菌及肠杆菌科细菌被列为严重耐药性[1]。据统计,2019年耐药细菌直接造成全世界127万人的死亡,多达495万人的死亡与耐药细菌感染有关,耐药性已成为人类第三大死亡原因[2]。随着AMR的进一步发展,MDR细菌导致的死亡人数还会持续增加,如果这一问题无法妥善解决,至2050年死亡人数预计高达 1 000 万[3]。
根据我国细菌耐药监测网(China Antimicrobial Resistance Surveillance System,CARSS)数据显示,2020年耐甲氧西林金黄色葡萄球菌(MRSA)的分离率达29.4%;大肠埃希菌对三代头孢菌素的全国平均耐药率为51.6%,对喹诺酮类抗生素的全国平均耐药率为50.7%;肺炎克雷伯菌对三代头孢菌素的全国平均耐药率为31.1%,对碳青霉烯类抗生素的全国平均耐药率为10.9%;肺炎链球菌对红霉素、克林霉素的全国平均耐药率高达90%以上;而鲍曼不动杆菌和铜绿假单胞菌对碳青霉烯类抗生素的全国平均耐药率分别为53.7%和18.3%[4]。
AMR对社会经济的负面影响难以估量。据美国疾病控制与预防中心估计,美国每年因耐药造成的经济损失高达550亿美元[5]。AMR已成为21世纪严重危害人群公共健康、影响人类经济生活的重大问题[6]。新型抗生素的研发速度远赶不上细菌对其产生耐药性的速度,投入大量人力和物力不断地研发新型抗生素已无法彻底解决日益严重的细菌耐药问题。因此,必须深入研究AMR的具体机制,同时积极探索应对AMR的新型防控策略。本文就AMR的生化机制及其防控策略进行总结和归纳,旨在为解决细菌耐药性问题的相关研究提供理论依据。
1 细菌耐药的机制
抗生素的杀菌机制如表1所示,主要包括:(1)抑制细菌细胞壁的合成;(2)改变细菌细胞膜的通透性;(3)抑制细菌蛋白质的合成;(4)抑制细菌核酸的合成;(5)抑制细菌的代谢途径。细菌耐药的类型和机制复杂,主要分为4类:(1)药物渗透障碍;(2)药物作用靶位改变;(3)药物失活;(4)主动外排(如图1和表2所示)[7]。其中由于革兰氏阴性菌外膜中含有脂多糖,革兰氏阴性菌适用上述4种机制,而革兰氏阳性菌不适用药物渗透障碍(外膜中没有脂多糖)和药物外排机制[6]。

表1 抗生素的杀菌作用机制Table 1 Action mechanisms of antibiotics

表2 细菌耐药的类型和机制Table 2 Types and mechanisms of antimicrobial resistance
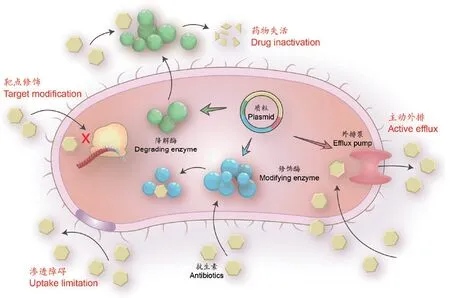
图1 细菌耐药机制Fig. 1 Mechanisms of antimicrobial resistance
1.1 药物渗透障碍
革兰氏阴性菌外膜中含有脂多糖形成的渗透性屏障,因此对某些抗生素的渗透性本质上低于革兰氏阳性菌。糖肽类抗生素,如万古霉素无法穿透外膜而对革兰氏阴性菌无效,证明了这种天然屏障的有效性。另外,不少抗生素必须穿透细菌外膜和细胞质膜作用其靶位才能发挥抗菌作用,细菌通过降低摄取来阻止抗生素分子抵达其胞内或胞浆周质的作用靶位导致耐药[8]。
1.1.1 外膜孔蛋白改变 外膜孔蛋白(outer membrane protein,Omp)是抗生素进入细菌体内发挥作用的主要通道,外膜孔蛋白量的减少或孔径减少及细胞壁增厚等均会降低外膜的通透性,使药物不易进入细菌抵达靶部位。既往研究认为细菌通过两种孔蛋白的改变来限制抗生素的摄取:(1)孔蛋白的缺失或严重减少;(2)突变致使孔蛋白选择性或功能的改变[9]。不同细菌通过不同的孔蛋白改变来发挥耐药作用,如肺炎克雷伯菌OmpK35、OmpK36、LamB和PhoE的缺失,OmpK36的突变导致其对碳青霉烯类抗生素耐药[10];耐碳青霉烯类抗生素的大肠埃希菌表现为OmpC和OmpF缺失,OmpC突变[11],OmpF缺失会导致β-内酰胺类抗生素、四环素类抗生素、喹诺酮类抗生素和氯霉素等药物的多重耐药;铜绿假单胞菌编码OprD2基因可发生多点突变、缺失突变和插入突变导致OprD2发生结构变化或缺失,使抗生素不能通过其细胞外膜,显著降低其对碳青霉烯类抗生素的敏感性[12]。
1.1.2 生物被膜 细菌在定植中,另一种降低抗生素摄取的机制是形成生物被膜(biofilm)。生物被膜是附着于物体表面被细菌胞外大分子包覆的有组织的细菌群体。其基质含有胞外多糖、蛋白质和细胞外DNA[13],能够削弱抗生素的渗透,同时为抵抗宿主免疫细胞的吞噬提供屏障[14]。研究表明,生物被膜对抗生素具有很强的抗性,生物被膜状态下的细菌可耐受的MIC浓度比浮游状态的细菌高1 000-2 000倍[15]。生物被膜的主要耐药机制包括:(1)降低抗生素在生物被膜中的渗透作用;(2)降低细菌自身新陈代谢和生长速率抵御抗生素的杀伤作用;(3)通过压力应答使生物被膜发生生理学改变,防止菌体受到抗生素的破坏作用等[16]。
1.2 作用靶位突变
细菌通过改变抗生素作用靶位的结构来降低抗生素与靶位的亲和力,引起耐药性。靶位改变通常是由染色体上细菌基因的自发突变引起的,由于抗生素与靶分子的相互作用通常具有相当的特异性,因此靶分子的微小改变可对抗生素结合产生重要影响[7]。靶位突变的经典机制是喹诺酮耐药决定区(quinolone resistance-determining regions,QRDR)突变。QRDR位于大肠埃希氏菌编码DNA促旋酶GyrA基因的67-106位氨基酸,GyrB基因的426-447位氨基酸之间;编码DNA拓扑异构酶Ⅳ ParC基因的63-102位氨基酸,ParE基因的420-441位氨基酸之间。DNA促旋酶与DNA拓扑异构酶Ⅳ中任何单个氨基酸发生突变均会削弱酶与喹诺酮类抗生素的相互作用,从而增强耐药性[17-18]。青霉素结合蛋白(penicillin-binding proteins,PBPs)是β-内酰胺类抗生素的作用靶点。细菌通过改变PBPs的结构(如金黄色葡萄球菌获得mecA基因后表达PBP2a)降低与β-内酰胺类抗生素的亲合力,从而对其耐药[19]。红霉素核糖体甲基化酶基因家族可使16S rRNA甲基化,并改变药物结合位点,阻断链球菌素、大环内酯类及林可胺类抗生素的结合[20]。耐万古霉素肠球菌(VRE)和金黄色葡萄球菌(VRSA)通过获得van基因使其肽聚糖末端D-Ala-D-Ala二肽结构变异为D-Ala-D-Lac,致使它们对万古霉素的亲和力降低1 000倍[21]。针对核糖体亚基抗生素耐药的产生可能与核糖体突变、erm基因参与的核糖体亚基甲基化以及核糖体保护有关,这些机制干扰了抗生素与核糖体的结合能力[7]。
多黏菌素(polymyxins)是一类包括5种不同化学结构(A-E)的阳离子环状多肽类抗生素,是常用于治疗产碳青霉烯酶等MDR菌株的最后一道防线之一。其作用机制是与革兰氏阴性菌外膜带负电脂多糖上的脂质A相互作用,取代细胞膜赖以稳定的Mg2+和Ca2+,引起细胞膜结构紊乱,细胞膜渗透性发生改变,细胞内物质渗漏,最终使细菌裂解死亡。细菌对多黏菌素耐药的主要机制是二元调控系统pmrAB和phoPQ基因突变、mgrB基因的完全缺失以及CrrB蛋白中的氨基酸替换上调了pmrC、pmrE和pmrHFIJKLM基因表达,使其编码的脂多糖修饰酶量增加,促进脂多糖修饰结合阳离子磷酸乙醇胺和4-氨基-4-脱氧-L-阿拉伯糖,从而产生耐药性[22]。另外,肺炎克雷伯菌荚膜多糖的过量生成使多黏菌素无法到达细菌外膜上的作用靶点[23]。铜绿假单胞菌还存在2个可能增加脂多糖中阳离子4-氨基-4-脱氧-L-阿拉伯糖表达的系统(ColRS和CprRS),可使外膜蛋白OprH过度表达,占据多黏菌素与脂多糖的结合位点[22]。鲍曼不动杆菌通过插入ISAba11和ISAba125等序列导致编码脂多糖的lpxA、lpxC和lpxD基因不表达,失去脂多糖的细菌无法与多黏菌素结合从而对多黏菌素耐药[24]。
2015年以前多黏菌素耐药被认为是染色体基因改变所致,而非质粒传播。同年11月我国研究人员发现了一个多黏菌素耐药的新基因mcr-1,其编码的内膜蛋白磷酸乙醇胺转移酶MCR能固定于细胞内膜并改变细胞膜构象,对脂质A基团进行化学修饰,包括对脂质A进行L-氨基-阿拉伯糖修饰以及将磷酸乙醇胺结合到脂质A的1-4’-磷酸基团上,使得细菌表面所带负电荷减少,削弱了多黏菌素和脂多糖之间的相互作用,导致多黏菌素耐药的产生(图2)[25-29]。由于mcr基因有较强的传播性以及能与不同耐药基因整合在同一个质粒上,因此mcr基因的报道越来越多。截至目前,世界范围内已经报道了mcr-1-mcr-10及其亚型基因[30]。在欧洲分离出的肺炎克雷伯菌中mcr-9基因检出率很高,同时伴有碳青霉烯酶基因,提示mcr基因可能已在欧洲人类来源的肺炎克雷伯菌分离株中广泛传播[31]。
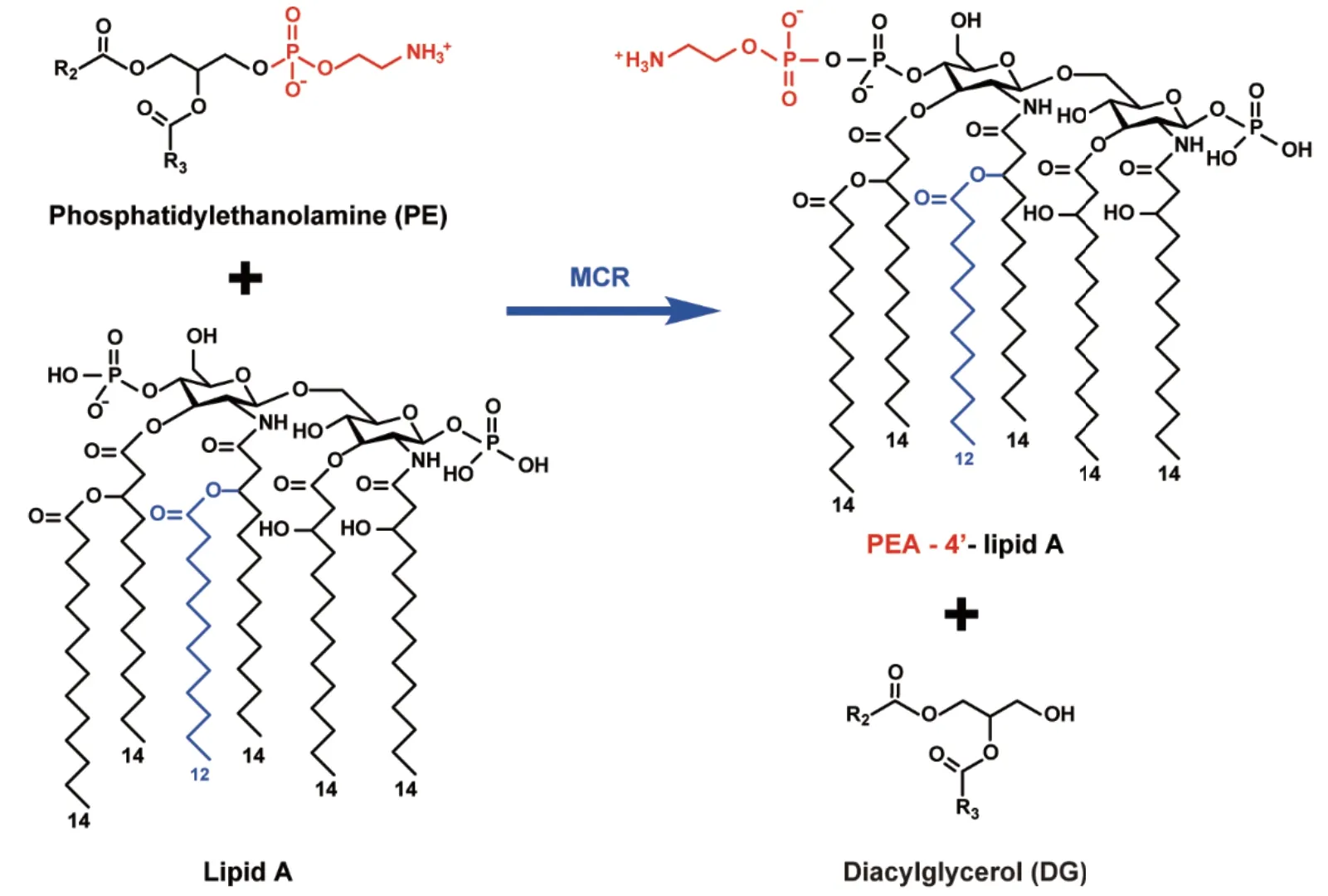
图2 MCR催化细菌脂质A的磷酸乙醇胺修饰(改自文献[26])Fig. 2 Phosphoethanolamine modification to lipid A catalyzed by MCR(Modified based on the reference[26])
1.3 药物失活
抗生素失活是细菌的最主要耐药机制。细菌主要通过两种方式使抗生素失去活性。(1)产生蛋白酶降解抗生素。目前细菌产生的灭活酶或钝化酶主要有β-内酰胺酶、氯霉素乙酰转移酶、氨基糖苷钝化酶和大环内酯酶-林克霉素类-链阳菌素类抗菌药物钝化酶。大肠埃希菌、铜绿假单胞菌、鲍曼不动杆菌等通过产生一种核黄素依赖单加氧酶Tet(X)使替加环素降解失活[32-33]。而微杆菌属细菌能够产生磺胺单加氧酶(SadA)及黄素还原酶(SadC)导致磺胺类抗生素发生ipso-羟基化及裂解[34]。肠杆菌科细菌通过产生糖苷酶、酯酶及磷酸化酶有效破坏大环内酯类抗生素的结构[35]。在细菌耐药机制中,对β-内酰胺类抗生素耐药的主要原因之一是产生β-内酰胺酶。β-内酰胺酶通过水解这类抗生素的β-内酰胺环(图3),抑制其与PBPs的结合而产生耐药。目前已发现近5 000种β-内酰胺酶,根据分子生物学分类法(Ambler分类法)可分为A、B、C、D四类,根据功能分类法(Bush分类法)可分为1类、2类和3类(表3)[36]。近年来产超广谱β-内酰胺酶(ESBL)的耐药细菌对人类健康构成严重威胁。ESBL是一种主要由肠杆菌科细菌产生,质粒编码合成的丝氨酸蛋白酶。常见的ESBL包括TEM、SHV、OXA、CTX-M等。另外,肠杆菌科细菌中还存在对碳青霉烯类抗生素具有活性的碳青霉烯酶。目前存在两种碳青霉烯酶:① 肺炎克雷伯菌碳青霉烯酶(KPCs),为A类丝氨酸β-内酰胺酶,能水解所有的β-内酰胺类抗生素,也能被β-内酰胺酶抑制剂影响[37];② 碳青霉烯耐药肠杆菌科酶(CREs),为B类金属β-内酰胺酶,能水解所有的β-内酰胺类抗生素,但不能被β-内酰胺酶抑制剂所抑制。常见的CREs包括IMP-1和VIM-1,由产CREs菌株导致的医院内感染的死亡率达到70%以上[38-39]。在2010年印度多个城市发现的产新德里金属β-内酰胺酶(NDM)的多重耐药细菌更是引起世界范围的恐慌[40]。
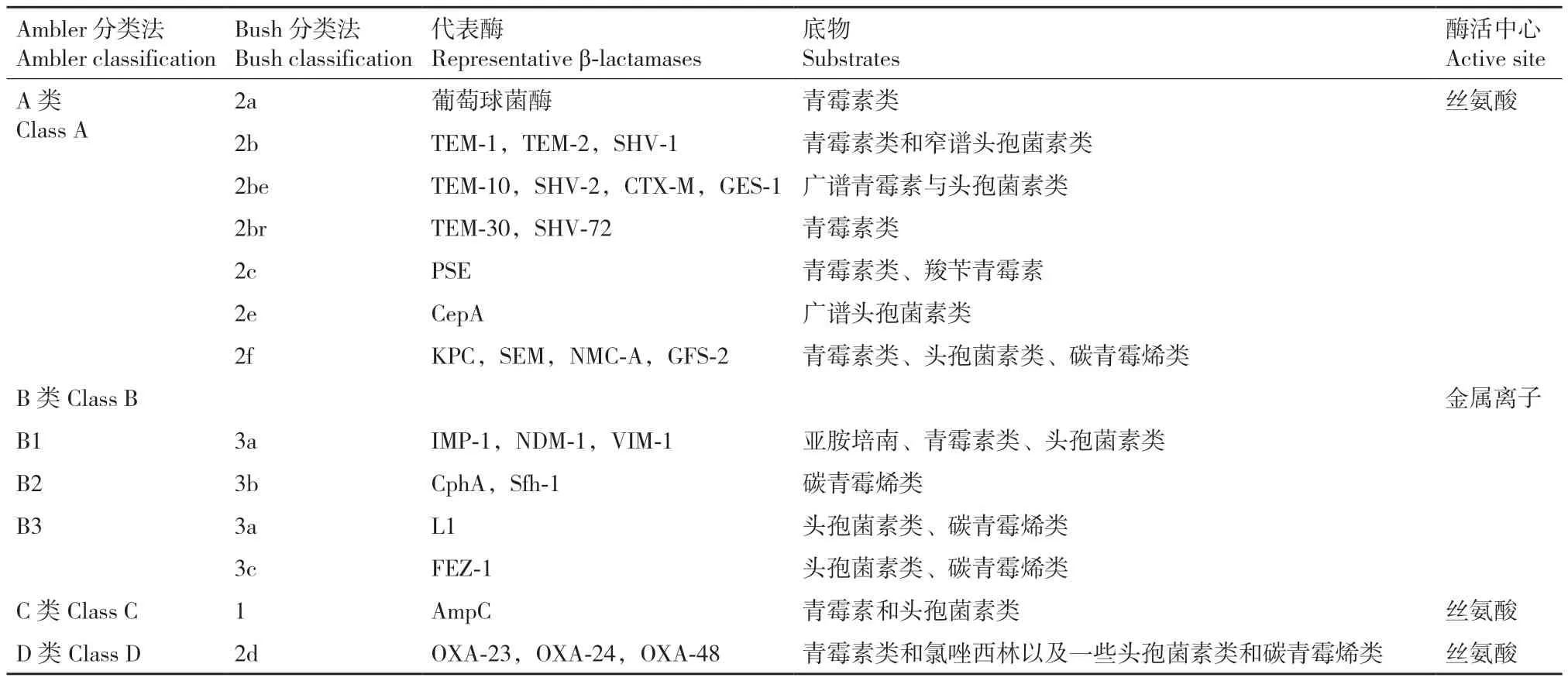
表3 主要β-内酰胺酶及分类Table 3 β-lactamases and their classification
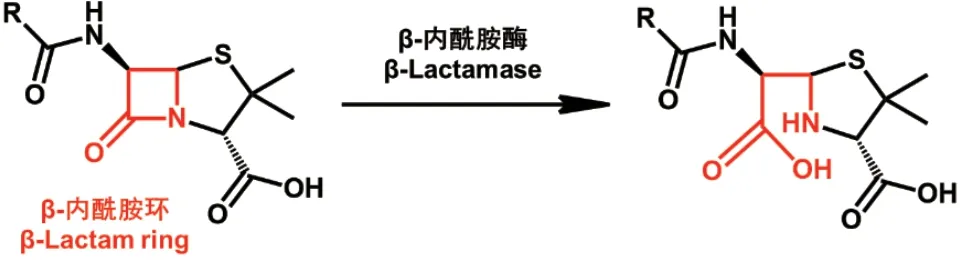
图3 β-内酰胺酶水解β-内酰胺类抗生素Fig. 3 Hydrolysis of β-lactams by β-lactamase
(2)添加或取代活性基团使抗生素失活。细菌通过产生能将多种活性基团(如乙酰基、磷酰基和腺苷基)连接到抗生素上的酶,阻止抗生素与作用靶位结合,如氨基糖苷类修饰酶(磷酸/乙酰/核苷酸转移酶)通过共价结合改变了氨基糖苷类抗生素结构上的氨基或羟基,导致其不能与作用靶位结合产生耐药性[6];氯霉素乙酰转移酶(CAT)催化乙酰辅酶A(CoA)依赖位于C-3位的羟基乙酰化,导致氯霉素修饰生成1-乙酰氯霉素,使其不能与细菌核糖体结合而耐药。
1.4 主动外排
主动外排系统的过量表达是细菌具有多重耐药性的重要原因之一。细菌利用通用或特异的外排系统将胞内的抗生素排出胞外,降低胞内抗生素浓度而产生耐药性。主动外排系统由外排蛋白、融合蛋白和外膜通道蛋白三部分构成。根据结构和能量供应进行分类,细菌的外排系统包括ATP结合盒(ABC)家族、主要易化子超家族(MFS)、小多药耐药(SMR)家族、耐药结节化细胞分化(RND)家族和多药及毒性化合物外排(MATE)家族(图4)[7]。其中RND家族是多组分泵,能与细胞周质膜融合蛋白和外膜蛋白协同将底物外排至细胞膜和细胞质外,其他外排系统均为单泵,穿过细胞质膜转运底物。四环素耐药是外排泵介导耐药的典型示例,铜绿假单胞菌的MexAB-OprM和肠杆菌科细菌的AcrAB-TolC能排出四环素[41]。鲍曼不动杆菌中AdeABC外排系统的高水平表达与美罗培南耐药密切相关[42]。
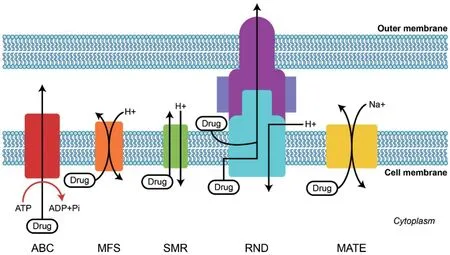
图4 外排泵家族的基本结构(改自文献[7])Fig. 4 General structure of efflux pump families(Modified based on the reference[7])
2 细菌耐药性防控新策略
2.1 抗菌肽疗法
抗菌肽(antimicrobial peptides,AMPs)是一类具有抑制或杀灭病原微生物的小分子多肽或小分子蛋白的总称,常含有10-50个氨基酸,相对分子量约为4 000 Da,带正电荷并具有较好的水溶性。抗菌肽根据其结构特征可分为α-螺旋型抗菌肽、β-片状抗菌肽、具有β发夹或环的抗菌肽、无规则卷曲的抗菌肽、线性结构抗菌肽、杂合抗菌肽等。抗菌肽的抗菌机制主要包括以下几个方面:(1)引起细胞壁损伤,抗菌肽通过与细胞壁合成前体分子结合、或干扰肽聚糖合成、或与肽聚糖结合等方式造成细菌细胞壁损伤,如Teixobactin与肽聚糖前体Lipid II和坦酸前体Lipid III结合抑制细菌细胞壁合成[43];(2)引起细胞膜损伤,抗菌肽与革兰氏阴性菌细胞外膜脂多糖结合形成跨膜通道破坏细胞膜完整性,与革兰氏阳性菌表面坦酸结合并附着于细菌表面,通过两亲性结构自聚形成构象簇穿过细胞膜并造成其受损,如带正电荷的抗菌肽LL-37与带负电荷的细菌结合,插入细菌细胞膜导致其内容物流出而死亡[44];(3)影响生物大分子合成及代谢关键酶活性,如抗菌肽P7通过抑制DNA复制和RNA合成引起大肠埃希菌的死亡[45];(4)引起免疫调节抑菌机制,抗菌肽在亚治疗剂量下可作为免疫效应分子调动并激活机体免疫细胞抑制或消除炎症,如LL-37可直接上调单核细胞趋化蛋白-1和IL-8的表达,诱导单核细胞产生大量IL-1β,间接诱导白细胞介素和单核细胞趋化蛋白质表达,或上调趋化因子受体CXCR-4、CCR2和IL-8RB表达,招募免疫细胞抵达感染部位[44]。抗菌肽抗菌作用机制独特,可单独或与抗生素联用,对革兰氏阳性和阴性多重耐药细菌具有广谱抗菌活性,是一类极具潜力的抗菌药物。目前全球范围内进入临床试验的抗菌肽不到50种,已上市的抗菌肽药物有阿尼芬净(anidulafungin)、杆菌肽(bacitracin)、卡泊芬净(caspofungin)、达托霉素(daptommycin)等。抗菌肽必须克服一些缺点(包括稳定性差、易被蛋白酶水解、生理条件下活性低及生产成本高等)后才能进入市场,对抗菌肽的结构进行优化来提高其生物活性、延长半衰期、降低细胞毒性等已成为当前抗菌肽领域的研究热点。
2.2 免疫疗法
基于主动免疫和被动免疫的疫苗和抗体的产生为预防和治疗多重耐药菌的感染提供了新思路。免疫疗法有助于打破抗生素耐药-新抗生素研发-新抗生素耐药的死循环,从根本上解决耐药细菌感染及耐药菌株流行的问题[46]。目前所研究的耐药细菌疫苗主要包括全菌灭活或减毒疫苗、外膜囊泡、重组DNA或蛋白亚单位疫苗、荚膜多糖疫苗。我国自主研发的耐药细菌疫苗重组金黄色葡萄球菌疫苗已获国家食品药品监督管理总局批准III期临床研究,该疫苗是我国首个自主研发的耐药细菌疫苗,其免疫攻毒保护率显著高于国际同类疫苗。德国Behringwerke公司和韩国Cheil Jedang公司开发的多个铜绿假单胞菌多糖蛋白结合疫苗也先后开展了人体临床研究,结果显示疫苗在健康志愿者中具有良好的安全性和较强的免疫原性,并能有效预防烧伤患者发生铜绿假单胞菌性菌血症[47]。另一种应对耐药细菌的策略是利用单克隆抗体,其主要作用是中和毒力因子的活性和抑制补体介导的细菌裂解。目前大约有10余种单克隆抗体处于临床试验阶段,有3种抗菌单克隆抗体药物已被批准上市,如Raxibacumab和Obiltoxaximab被批准用于预防和治疗吸入性炭疽。
2.3 噬菌体疗法
噬菌体是一类能够特异寄生在细菌、放线菌、真菌等微生物内的DNA或RNA病毒,根据对宿主菌的作用方式不同可分为烈性噬菌体(溶菌型噬菌体)和温和噬菌体(溶原型噬菌体)。烈性噬菌体与细菌表面特定受体结合并将其遗传物质注入宿主内进行复制,子代噬菌体在细胞溶解酶和穿孔蛋白的作用下裂解细菌后导致感染终止,并释放到周边环境中继续重复杀伤过程[48]。噬菌体治疗的优势在于研发周期短、成本低且特异性感染宿主菌。特异性使噬菌体在治疗过程中对患者正常菌群没有影响,无抗菌药物的毒副作用。噬菌体的抗菌机制独特,能自我复制并增强抗菌疗效,对于耐药细菌感染或抗生素治疗无效的患者具有天然的优势。但单一噬菌体抗菌谱窄,能引起机体免疫应答不足等,目前主要采取噬菌体鸡尾酒疗法、抗生素与噬菌体联用、噬菌体裂解酶与抗菌制剂联用等方法来提高疗效[49]。2020 年 Tkhilaishvili团队[50]报道了一例多重耐药铜绿假单胞菌引起的假体关节周围感染,在抗生素和手术治疗均失败的情况下,患者接受噬菌体辅助治疗,发现噬菌体与抗生素联用可有效消除感染,在治疗第3天的引流液中未分离到铜绿假单胞菌。2021年该团队[51]再次报道了一例噬菌体与抗生素联用成功治疗心室辅助装置感染的案例。在手术前静脉施用噬菌体共3次,并局部应用噬菌体,手术清创期间再次递送噬菌体与抗生素,手术结束后并未分离出多重耐药铜绿假单胞菌。李莉莎等[52]尝试使用噬菌体雾化吸入治疗泛耐药肺炎克雷伯菌引起的肺部感染,发现分离株的噬菌体裂解谱发生了变化,患者临床症状改善。
2.4 抗菌光动力疗法
抗菌光动力疗法(antimicrobial photodynamic therapy,aPDT)是一种利用无毒的光敏剂(PS)、特定波长的可见或近红外光以及耐药细菌周边或内部的分子氧产生光毒性反应来杀伤耐药细菌的治疗模式[53]。如图5所示,PS在特定波长光的照射下从基态经过寿命极短的激发单重态,系间窜越到激发三重态。三重态的PS与分子氧通过I型反应产生超氧阴离子自由基(O2·-)、羟基自由基(·OH)、过氧化氢(H2O2),或通过II型反应产生单线态氧(1O2)[54]。这些活性氧(ROS)对耐药细菌的生物大分子和细胞结构造成不可逆转的氧化损伤,导致耐药细菌的死亡[55]。aPDT对几乎所有的耐药细菌都具有良好的杀菌活性[56],由于ROS非特异性的氧化作用模式,氧化损伤发生在细菌内许多作用靶点(如核酸、脂质和蛋白质等)和细胞结构,一般认为耐药细菌不太可能对aPDT产生抗性机制[57]。例如本团队[58]利用20个重复周期的亚致死剂量aPDT处理多重耐药大肠埃希菌后发现,细菌没有对aPDT产生抗性,而且对头孢他啶和多黏菌素E的MIC值分别降低了4倍和2倍。aPDT是一种独立于抗生素的杀菌模式,对宿主损伤小,多次治疗不会诱导耐药,因此被认为是很有前途的治疗细菌感染方法之一,尤其是多重耐药细菌引起的感染。目前aPDT已被用于部分浅表或局部耐药细菌感染的治疗,但治疗深度有限这一问题仍需解决。
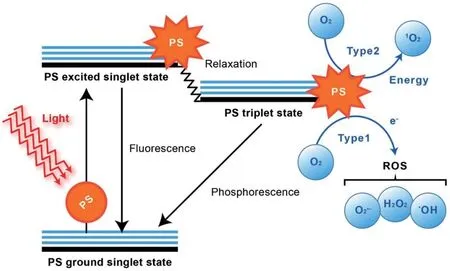
图5 光敏剂的贾布朗斯基图(改自文献[54])Fig. 5 Jablonski diagram of photosensitizer(Modified based on the reference[54])
2.5 益生菌疗法
益生菌(probiotics)是定植于肠道、生殖系统内能产生确切健康功效的具有活性的有益微生物的总称。其合成的活性短肽——细菌素,能抑制部分人体致病菌、调节肠道菌群以及帮助益生菌定植[59]。细菌素的抑菌机制有两种:(1)破坏细菌细胞壁使内容物流出,主要针对革兰氏阳性菌;(2)进入细菌胞内抑制基因或蛋白的表达,主要针对革兰氏阴性菌。细菌素中的羊毛硫肽(lanthipeptides)是目前益生菌抑菌领域研究最深入的一种,乳酸链球菌素(nisin)是羊毛硫肽的主要代表[60]。羊毛硫肽除了能抑制一般细菌外,还对部分耐药细菌具有抑制作用,如MRSA和VRE等。研究表明加氏乳杆菌OLL2716对耐药幽门螺杆菌在体内外的生长及定植均具有明显的抑制作用[61]。Matthew Chang团队[62]发现经过基因改造的益生大肠埃希菌Nissle 1917能在动物模型中预防和消除铜绿假单胞菌引起的肠道感染。目前益生菌抗耐药细菌的研究尚处于体外试验阶段,未来需要进一步开展体内和临床试验。
2.6 抗菌纳米颗粒技术
近年来抗菌纳米颗粒技术成为新的对抗耐药细菌研究热点。常见的抗菌纳米颗粒主要有银纳米颗粒(silver nanoparticles,AgNPs)、金属氧化物纳米颗粒、光热转换纳米颗粒、抗生素共轭纳米颗粒等[63],它们通过破坏细菌细胞膜、与细菌DNA或蛋白质相互作用、间接启动ROS的产生等方式实现抗菌作用。AgNPs是最常见的抗菌纳米颗粒,具有广谱的抗菌活性,其作用机制包括:(1)干预细菌细胞壁或细胞膜的合成;(2)干扰细菌能量代谢;(3)抑制细菌关键酶的活性;(4)抑制细菌DNA的合成;(5)产生ROS等。纳米颗粒的细胞毒性是一个不容忽视的缺点。在抑菌浓度下,AgNPs被证实具有一定的细胞毒性。为了克服这个问题,国内外学者发展了羧甲基罗望子多糖、壳聚糖、阿拉伯胶、聚乙烯吡咯烷酮以及二氧化钛封端的AgNPs,它们在保留抗菌活性的同时能显著降低细胞毒性[64]。
2.7 反义寡核苷酸和基因编辑技术
反义寡核苷酸是与细菌mRNA互补的RNA分子,二者特异性结合后能抑制mRNA的翻译,从而抑制相关基因的表达,在基因水平调控的分子药物。反义寡核苷酸包括反义DNA和反义RNA,其机制涉及顺式和反式序列,顺式反义序列一般可以在mRNA附近的调节区域找到,或从同一遗传位点的互补链转录而来;反式反义序列从较远的遗传位点转录而来。反义寡核苷酸能够抑制细菌耐药酶的翻译、也可用于发现新的抗菌物质、开发高度敏感的抗菌屏障及作用模式[65-66]。罗晓星团队[67]利用反义技术抑制MRSA群体感应(quorum sensing,QS)系统中关键分子agrA的表达,阻断其信号通路并降低细菌的致病力,为抗MRSA感染提供了一种新思路。
由于抗生素的大量使用,在动物和人体内诱导出抗生素耐药基因,这些基因进入环境后,通过横向转移机制诱导耐药菌的产生。成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/ Cas系统作为一种细菌适应性免疫系统,被认为是控制耐药菌株的新策略之一。该系统用于对抗细菌基因组序列的可编程Cas核酸酶,也有望降低细菌对抗生素的耐药性。根据效应蛋白的数量,CRISPR / Cas系统可分为I型和II型两大类,作用机制可分为3个阶段:新间隔序列的获得与整合(适应)、cr RNA和Cas蛋白的生成(表达)以及cr RNA引导的核酸靶向切割(干扰)。CRISPR / Cas系统可以区分致病菌和共生菌,并能选择性的剔除耐药基因,或通过消除含有耐药基因的质粒使细菌重新对抗生素敏感[65]。在所有Cas蛋白中,针对耐药细菌最有前景的为CRISPR-Cas9、dCas9、nSpCas9 和 Cas13a。Kim 等[68]利用 CRISPR /Cas9 系统靶向大肠埃希菌中的抗生素耐药基因,成功清除了产ESBL大肠埃希菌内的抗生素耐药质粒。Citorik 等[69]和 Bikard 等[70]分别以大肠埃希菌和金黄色葡萄球菌内的抗生素耐药质粒为靶标,利用噬菌体为载体传递CRISPR / Cas9系统,最终细菌中的抗生素耐药质粒被成功清除,耐药菌对抗生素的敏感性得到恢复。目前,亟需开展体内研究来解决该系统存在的不足,包括如何通过噬菌体、质粒以及纳米颗粒将CRISPR-Cas系统高效输送至细菌体内的递送问题,以及宿主基因组中潜在脱靶修饰的副作用等[71]。
2.8 其他新策略
应对AMR的其他新策略包括宿主导向疗法(host-directed therapy,HDT)、毒力因子(virulence factors,VF)抑制剂、QS抑制剂、纳米酶等。细胞内的病原菌能够与宿主共同进化,操纵宿主因子继而维持病原菌的生存。在这种情况下,HDT可能是一种很有前景的抗生素替代疗法,它能靶向宿主因子并消除细胞内病原菌的复制,同时引起宿主针对病原菌的免疫反应,并通过增强病理部位的愈合来减少慢性炎症。HDT靶向病原菌生命周期并有助于根除病原菌,这一特性契合了耐药细菌感染的治疗[72-73]。VF抑制剂是近年来的研究热点之一,由于它并不直接抑制细菌的生长,因此大大降低了耐药细菌出现的概率,同时也降低了菌株的致病力[74]。许多细菌的行为受到QS的调节,QS依赖细菌之间可扩散的信号分子进行通信,能够调节细菌代谢、毒力和生物被膜形成等生理过程。群体淬灭是通过化学或酶的手段抑制QS,对抗QS调节从而达到抗菌效果。QS抑制剂是目前研究最多的一类物质,它能抑制QS调控的功能达到抗菌目的[75]。目前已发现了几种QS抑制剂,包括假单胞菌喹诺酮信号通路阻滞剂、丁子香酚、阿司匹林、布洛芬及吲哚美酰亚胺等[76]。纳米酶是新一代人工模拟酶,包括过氧化物酶、氧化酶、卤代过氧化物酶和脱氧核糖核酸酶等,它兼具纳米材料独特的理化性质和类酶催化活性,相较于天然抗菌酶(溶菌酶、髓过氧化物酶等)具有结构稳定、抗菌谱广、生产成本低等优点[77]。近年来新型抗菌策略层出不穷,未来将被更加深入地研究及进一步转化。
3 总结与展望
抗生素的发现是现代医学史上的里程碑,使人类告别无法控制细菌感染的“黑暗时代”。但抗生素的过度使用和滥用已导致细菌耐药率持续升高,耐药程度愈发严重,多重耐药甚至全耐药菌株普遍出现。目前,多重耐药细菌引起的感染已成为临床亟待解决的医学难题,使用单一抗生素已无法达到预期治疗效果。而新型抗生素的研发速度远落后于细菌耐药的发展速度,投入大量人力、物力研发抗生素又会陷入抗生素耐药-新抗生素研发-新抗生素耐药的死循环,人类正在迈入“后抗生素时代”。为了应对日益严重的细菌耐药性问题,在加强耐药细菌监测力度、减少抗生素使用的同时,亟需深入开展细菌耐药生化机制的研究。现有的研究表明,细菌耐药生化机制具有复杂性和多样性,包括:外膜孔蛋白量减少或孔径减少、形成生物被膜减少抗生素摄取的药物渗透障碍机制;通过改变抗生素作用靶位的结构来降低抗生素与靶位亲和力的作用靶位突变机制;产生水解酶、添加或取代活性基团使抗生素失活的药物失活机制;以及利用外排系统将胞内抗生素排出胞外、降低胞内抗生素浓度的药物外排机制。此外,仍需积极探索和发展细菌耐药性防控新策略或新方法。尽管抗菌肽疗法、免疫疗法、噬菌体疗法、抗菌光动力疗法、益生菌疗法、抗菌纳米颗粒技术、反义寡核苷酸和基因编辑技术、宿主导向疗法等在防控耐药细菌方面显现出巨大的应用潜力,其中一些方法或技术已用于耐药细菌感染的治疗,但仍存在不少的问题需要深入研究和探索。相信在不久的将来,细菌耐药这一医学难题一定能够得到妥善地解决。
