NLRP3 炎症小体与神经病理性疼痛关系的研究进展 *
2022-11-01庞淼一马奕然王培培
庞淼一 马奕然 王培培 李 纤 杨 菲△
(1 首都医科大学基础医学院,北京 100069;2 首都医科大学基础医学院神经生物学系,北京 100069;3 首都医科大学基础医学院生物化学与分子生物学系,北京 100069)
国际疼痛学会(the International Association for the Study of Pain, IASP) 给神经病理性疼痛 (neuropathic pain, NP) 所下的定义为:由躯体感觉系统的损害或疾病所导致的疼痛。这种疼痛通常表现为自发痛、触诱发痛、痛觉过敏和感觉异常等临床特征[1],临床上以病程超过3 个月的慢性NP 病人居多。世界上6.9%~10%的人群经历过NP,法国和英国的流行病学调查显示NP 的患病率为7%~8%[2],国内则有9000 万NP 病人[1]。这些病人的心理健康和社会功能大多受到严重不良影响,导致病人生活质量下降,给个体、家庭和社会造成沉重负担。但与此同时,面对如此庞大的患病人群,现有的临床治疗方法无法有效缓解大部分NP 病人的临床症状,例如常用的阿片类药物、非甾体抗炎镇痛药等对于NP 的治疗效果都十分有限,此外还容易诱发多种不良反应[3]。目前,关于NP 的发病机制尚不完全明确,这也极大限制了NP 治疗方法的研发[4]。
既往的研究已经表明,NP 与外周和中枢神经系统的神经元敏化有关。炎症可以促进再生和愈合,但如果炎症的消退受到干扰,则有可能导致慢性疼痛的发生。除了神经元,NP 还涉及许多非神经元细胞,包括免疫细胞、神经胶质细胞等[5]。越来越多的研究表明,受损或病变神经部位发生的免疫反应激活了局部组织中的天然免疫细胞,这一过程在外周和中枢的神经元敏化中发挥了重要作用。随后炎症介质(如细胞因子、神经营养因子和趋化因子)的释放启动局部炎症反应,进而引发更广泛的级联效应[6]。神经炎症环境能够引起小胶质细胞和星形胶质细胞的激活,影响突触效能和疼痛敏感性,在诱发和维持NP 方面起关键性作用[7]。
炎症小体是一种能感知多种病原体或危险信号的多蛋白质复合物,能够调节胱天蛋白酶依赖性的炎症反应和细胞凋亡。目前发现的炎症小体主要包括NLRP1、NLRP3、NLRC4、IPAF 和AIM2, 其中NLRP3 炎症小体与NP 的关系最为密切。有研究表明,NLRP3 炎症小体在许多与痛觉相关的神经和免疫细胞中均存在表达(见表1),包括:①背根神经节中的神经元,脊髓背角中的神经元、小胶质细胞和星形胶质细胞[8];②与外周感觉神经系统以及周围微环境相关的炎症细胞(如巨噬细胞、中性粒细胞、肥大细胞等)[9];③痛觉相关脑区中的神经细胞(如大脑皮层的小胶质细胞等)[10]。本文将重点关注NLRP3 炎症小体在各种类型NP 相关疾病中的作用途径和机制,希望为探索NP 的发生发展机制和治疗前景提供思路。
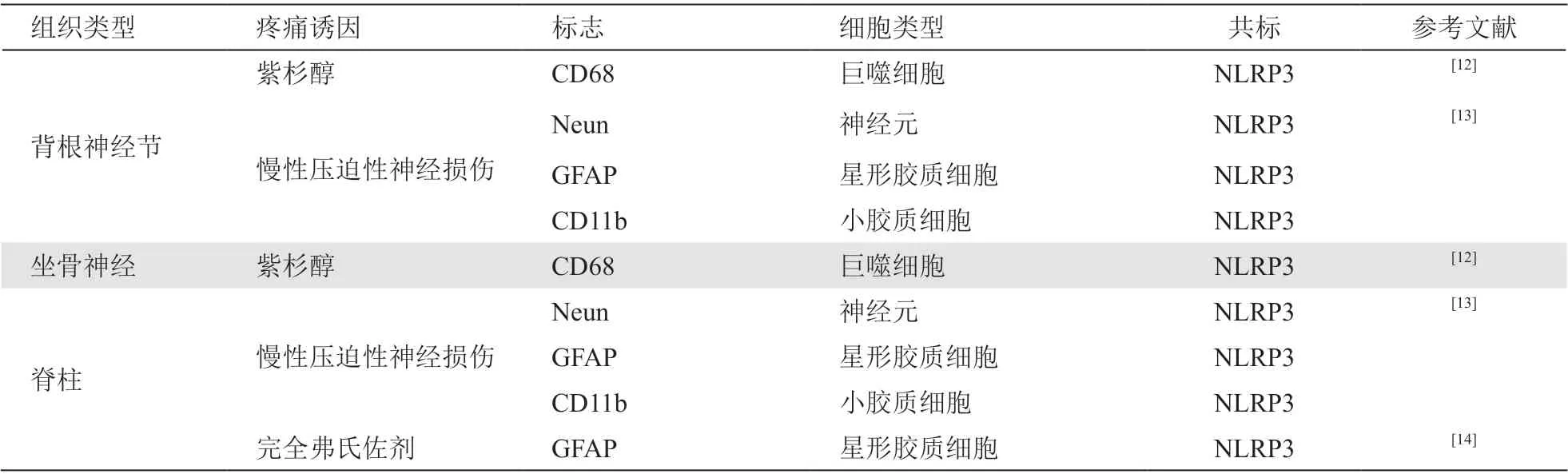
表1 NLRP3 炎症小体在不同诱因导致的疼痛中的表达位置
一、NLRP3 炎症小体的组成和功能
NLRP3 (NOD-like receptor protein 3)蛋白质由冷诱导自身炎症综合征基因1 (cold induced autoinflammatory syndrome 1, CIAS1) 编码。其蛋白质结构由3 个主要结构域组成:N 端吡咯结构域、中心核苷酸结合和寡聚化结构域以及C 末端富含亮氨酸重复序列结构域。NLRP3 炎症小体以NLR 作为受体蛋白、ASC 作为接头蛋白、Caspase 作为效应蛋白[11]。
NLRP3 炎症小体的直接激活信号可以是病原体相关分子模式和损伤相关分子模式,也可以是其他化学、物理和环境因素。研究表明,NLRP3 炎症小体的激活过程需要两种信号的参与。初始化(第一信号)通过激活NF-κB 通路,上调与炎症小体相关的蛋白(包括炎症小体传感器蛋白,IL-1β 和IL-18)的表达。外界刺激激活细胞产生第二信号,目前普遍认为主要有 3 种潜在刺激:细胞内K+浓度的降低、线粒体ROS 的产生和溶酶体膜的破坏[11]。刺激启动的激活组装(第二信号)进一步触发炎症小体传感蛋白与炎症小体接头蛋白的聚集,并招募Caspase-1。活化(水解)的Caspase-1 不仅能促使IL-1β 和IL-18成熟并分泌,并且还促使Gasdermin D (GSDMD) 切割,切割后的GSDMD 在细胞膜上形成孔洞,引发促炎性细胞死亡,即细胞焦亡[11]。
NLRP3 炎症小体在多种神经系统疾病中的作用已受到广泛关注,例如抑郁症病人外周血中NLRP3含量升高,在药物治疗后下降。在小鼠抑郁症模型中,海马中的NLRP3 炎症小体被激活,而敲除NLRP3可以阻断抑郁样行为。此外,在阿尔茨海默病模型动物中阻断IL-1β可以减缓脑中Tau蛋白过度磷酸化,拯救认知障碍,使用NLRP3 抑制剂后模型动物的淀粉样蛋白沉积减少,认知功能得到改善[6]。
目前已知的NLRP3 抑制剂有MCC950、CY-09、JC-124、Oridonin 等(见表2),其中在基础研究中最常使用的是MCC950。虽然大量临床前研究显示NLRP3 炎症小体抑制剂能够治疗多种神经系统疾病,但是目前还没有一种抑制剂能够在临床上应用。因此NLRP3 炎症小体与人类疾病的关系,以及能否作为有效的治疗靶点仍有待研究和证明。

表2 NLRP3 炎症小体抑制剂在动物疾病模型中的应用
二、NLRP3 炎症小体与慢性神经病理性疼痛
1. NLRP3 炎症小体在外周神经病理性疼痛中的研究
慢性周围神经病理性疼痛是指由于外周感觉神经系统损伤或疾病而导致的慢性疼痛[4],IASP 在2019 年提出将其分为以下6 类:三叉神经痛、周围神经损伤后的慢性病理性疼痛、痛性多发神经病变、痛性神经根病变、带状疱疹后神经痛和其他待明确及未明确的慢性周围神经病理性疼痛[23]。本文主要讨论前4 类与NLRP3 炎症小体之间的关系。
(1)NLRP3 炎症小体与三叉神经痛
三叉神经痛 (trigeminal neuralgia, TN) 是局限于三叉神经的一个或多个分支的口面部神经性疼痛。疼痛经常反复突然发作和停止,可由无害刺激引起,病人常将其描述为电击样、枪击样痛或刺痛,部分病人在阵发性疼痛之间还会经历持续性疼痛[23]。
在完全弗氏佐剂注射造成的小鼠TN 模型中,研究人员发现三叉神经结内NLRP3 炎症小体及相关蛋白的 mRNA 水平明显升高,血清中炎症小体相关蛋白的表达量也同时升高[24]。在另一项研究中,研究人员将牙植体错位移植压迫小鼠下牙槽神经建造TN 模型,发现造模后的0 到21 天,小鼠延髓背角内NLRP3 的mRNA 水平逐渐升高[25]。
随后研究人员试图通过抑制NLRP3 的表达改善TN 模型小鼠的面部机械痛敏,发现内源性非编码RNA (miR-186)过度表达虽然可以直接抑制NLRP3 的表达水平,但对于小鼠面部机械痛敏却没有明显的改善作用[24]。而在下牙槽神经受压迫的模型鼠中,研究人员敲低小鼠体内的 NLRP3 后发现小鼠的机械痛敏得到了缓解[25],提示直接敲低 NLRP3 的表达对于疼痛的缓解作用显著优于miR-186。
(2)NLRP3 炎症小体与周围神经损伤后的慢性神经病理性疼痛
周围神经损伤后的慢性神经病理性疼痛 (chronic neuropathic pain after peripheral nerve injury, CNP-pni)是由周围神经损伤引起的持续性或复发性疼痛。疼痛的具体病理表现和范围与所损伤的神经有关[23]。
现有研究发现在 CCI(慢性压迫性神经损伤)模型[13]、SNL(脊神经结扎)模型中[26],模型动物出现明显的机械痛敏和热痛敏,同时 NLRP3 在脊髓内的表达升高。在CCI 模型中,大鼠脊髓背角内 NF-κB (p65)表达逐渐升高,并且在14 天达到峰值。NF-κB 进而上调脊髓背角神经元及小胶质细胞中NLRP3 的表达(见表1),其下游Caspase-1 和炎症因子 IL-1β 的表达也上升,进而加重CCI 大鼠的机械痛敏和热痛敏[13]。NLRP3 抑制剂MCC950[15]和红景天苷[27](传统中草药成分)都被证实可以通过抑制该通路的激活减轻CCI 模型鼠的疼痛症状。临床抗炎药物芍药苷对于模型动物NP 的抑制也被证实有NLRP3 的参与,芍药苷可以同时增强Nrf2的核转位并抑制 NF-κB 的表达,通过这两条途径共同抑制NLRP3 的表达从而降低CCI 模型鼠的机械痛敏。但研究者同时指出Nrf2 的激活在芍药苷的作用中可能占比很小,因为抑制 Nrf2 的表达后芍药苷仍然有效[15]。而右美托咪定完全通过激活 Nrf2起到抑制 NLRP3 表达的作用[28]。上述研究提示TXNIP-NLRP3 途径在多个周围神经损伤模型中导致了疼痛的发生,很有可能成为 CNP-pni 治疗药物新的作用通路,而Nrf2/NLRP3 通路是否能够有效改善 CNP-pni 仍然需要更进一步的研究证实。但在SNI(保留神经损伤)模型鼠体内,研究人员发现NLRP3 表达水平与小鼠的机械痛敏之间并不存在直接的联系。
(3)NLRP3 炎症小体与痛性多发神经病变
痛性多发神经病变 (painful polyneuropathy, PP)是由代谢性、自身免疫性、家族性或传染性疾病,以及暴露于环境、职业毒素或使用神经毒性药物治疗引起的多神经病所导致的慢性疼痛[23]。其中较为常见的是糖尿病引发的痛性多发神经病 (diabetic peripheral neuropathy, DPN)。研究者通常使用链脲佐菌素 (streptozotocin, STZ) 法构建 DPN 模型,实验发现该模型小鼠脊髓和背根神经节内 NLRP3 蛋白表达升高,并且使用三萜皂苷抑制 NLRP3 表达可使 STZ 小鼠的机械痛阈和热痛阈提高[29]。
多种药物都被证实通过抑制 NLRP3 在 DPN 模型中发挥镇痛作用。研究者发现高糖环境使细胞中的磷酸化 AMPK、TXNIP 和激活的 NLRP3 炎性小体增多。在使用红景天苷进行治疗后,DPN 小鼠背根神经节中前述三种因子减少并且小鼠的机械痛阈明显提高[30]。另一项研究则指出獐牙菜苦苷能缓解STZ 模型鼠触觉过敏症状,机制之一可能是抑制了大鼠脊髓内 NOXs/ROS/NLRP3 信号通路,从而使抑炎因子表达上升、促炎因子表达下降,达到纠正DPN 导致的炎性因子失衡的作用[31]。
除了DPN,另一种常见的痛性多发性神经病是化疗药物引发的外周神经病变 (chemotherapy induced peripheral neuropathy, CIPN)。紫杉醇作为一种常用的癌症化疗药物,可在动物模型中诱发CIPN。研究者证实紫杉醇和三磷酸腺苷 (ATP) 共同作用可以诱导大鼠线粒体损伤,使活性氧的产生增加,导致L4-6背根神经节和坐骨神经的巨噬细胞中NLRP3 炎症小体激活(见表1),从而引发大鼠机械痛敏。在使用ROS 抑制剂PBN (phenyl-N-tert-butylnitrone) 后,研究者发现被激活的NLRP3 炎症小体减少,同时大鼠机械痛敏得到改善[12]。研究认为,另一种常用化疗药物奥沙利铂可以通过降低脊髓中细胞外腺苷信号的表达诱导星形胶质细胞中ADK的表达,进而使 NLRP3(见表1)和 IL-1β 表达增加,诱发神经炎症导致疼痛发生[32]。后续给CIPN 大鼠使用 MCC950 抑制 NLRP3 表达后,大鼠的疼痛明显好转[32]。但另一组研究人员给小鼠分别注射奥沙利铂和紫杉醇建造CIPN 模型,检测发现这两种模型鼠脊髓内 NLRP3 相关炎症因子的表达水平都没有发生明显变化[33]。所以紫杉醇和奥沙利铂是否导致NLRP3 的水平变化还需要进一步的探索与研究。
(4)NLRP3 炎症小体与痛性神经根病变
痛性神经根病变 (painful radiculopathy, PR) 是由涉及颈、胸、腰或骶神经根的病变或疾病引起的持续性或复发性疼痛。其中,脊柱退行性改变(包括韧带和椎间盘)是最常见的病因。这种疼痛可以自发,但通常是由特定的姿势或在运动过程中诱发的[23]。
在临床上,腰椎间盘突出症 (lumber disc herniation,LDH) 和软骨终板退行性病变是常见的可以引起神经根痛的疾病。研究者取大鼠自体髓核移植到神经根附近对神经根进行压迫模拟LDH,检测发现手术组大鼠背根神经节内NLRP3 表达上调[34]。临床研究中获得的数据提示,在腰椎软骨终板退行性病变病人体内NLRP3、Caspase-1 和IL-1β 表达阳性的细胞数量增多,作者因此推测NLRP3/Caspase-1/IL-1β轴的激活可能引发椎间盘退行性病变和腰痛[35]。在临床前研究中,富勒醇纳米颗粒和MaR1 两种抗炎药物都被证实可以通过抑制NLRP3/Caspase-1/IL-1β轴来提高NP 移植模型鼠的机械痛阈[36]。
2. NLRP3 炎症小体与慢性中枢神经病理性疼痛
慢性中枢神经病理性疼痛是由中枢体感神经系统的病变或疾病引起的慢性疼痛。IASP 在2019 年提出将慢性中枢神经病理性疼痛分为5 种:脊髓损伤相关的慢性中枢神经病理性疼痛、脑损伤相关的慢性中枢神经病理性疼痛、卒中后慢性中枢性疼痛、多发性硬化症导致的慢性中枢神经病理性疼痛和其他明确的或未明确的慢性中枢神经病理性疼痛[14]。本文主要讨论前4 种与NLRP3 炎症小体之间的关系。
(1)NLRP3 炎症小体与中枢损伤相关的神经病理性疼痛
脊髓损伤相关的慢性中枢神经性疼痛(chronic central neuropathic pain associated with spinal cord injury, CCNP-sci) 是由脊髓体感通路的病变或其他疾病引发的症状。脊髓损伤包括外力损害或疾病导致的脊髓功能损伤。疼痛可能自发或由某些因素诱发,病人通常对疼痛刺激的反应增强(痛觉过敏)或对正常无疼痛刺激的疼痛反应(痛觉超敏)[23]。
研究者发现SCI 模型小鼠脊髓中NLRP3 的转录水平显著升高,并且在使用药物抑制了NLRP3的表达水平后,模型鼠的机械痛阈同步升高[37]。中重度胸椎挫伤的模型小鼠使用内源性酮体NLRP3抑制剂D-β-羟基丁酸酯 (DBHB) 治疗后,小鼠脊髓中NLRP3 炎症小体组分表达、炎症小体激活和炎症因子IL-1β、IL-18 的表达水平降低,小鼠的运动功能得到了明显改善,且脊髓损伤引起的痛觉超敏得到了缓解[38]。上述研究结果表明,NLRP3 炎症小体在脊髓中的表达水平、激活程度与SCI 诱导的中枢神经病理性疼痛呈正相关。
脑损伤相关的慢性中枢神经病理性疼痛 (chronic central neuropathic pain associated with brain injury,CCNP-bi) 是由躯体感觉皮层、连接的脑区或脑内相关通路的病变或疾病引起的。最常见的脑损伤是外伤性脑损伤 (traumatic brain injuries, TBI),由外力如直接撞击头部或头部被暴露在冲击波中导致。TBI常见的伴随症状之一就是慢性疼痛。在TBI 的动物模型中,神经炎症已被证明存在于许多与疼痛相关的大脑区域,包括丘脑、皮层、海马、杏仁核和前额叶皮质[39]。进一步的实验结果显示,NLRP3 炎症小体参与创伤性脑损伤后继发性脑损伤的发生,包括慢性痛症状等[40]。另一方面MCC950 被证实可以通过抑制NLRP3 炎症小体的激活,改善脑损伤模型小鼠的脑水肿和血脑屏障的损伤[41]。
(2)NLRP3 炎症小体与卒中后慢性中枢疼痛
卒中后慢性中枢性疼痛 (chronic central poststroke pain, CCPSP) 是由大脑或脑干缺血或出血引起的一种慢性神经病理性疼痛综合征[42]。在脑缺血再灌注后的小胶质细胞、神经元和内皮细胞中均检测到NLRP3 炎症小体激活[43],且脑缺血组的脑组织中NLRP3 炎症小体蛋白水平比对照组高4 倍。同时,缺血性中风后,NLRP3 激活可导致梗塞中心的神经元和小胶质细胞死亡[43]。
根据现有的研究,有学者提出了NLRP3 炎症小体在CCPSP 中可能的作用机制:脑卒中进展中无菌性炎症反应可诱导巨噬细胞、中性粒细胞、淋巴细胞等的浸润和活化,从而导致炎症和神经细胞死亡[44]。坏死细胞中线粒体的损伤产生ROS,激活NLRP3 炎症小体。在机制上,ROS 的产生使TXNIP从硫氧还蛋白中解离,继而TXNIP 与NLRP3 一起易位至线粒体,从而激活NLRP3 炎症小体[6]。
此外在脑损伤过程中, K+水平升高和氧化的线粒体DNA 能诱导NLRP3 激活并释放IL-1β 和IL-18,脑损伤过程中坏死的细胞释放IL-1β 和IL-18 可以导致无菌炎症反应,从而激活NLRP3。NLRP3、IL-1β、IL-18 进一步引发的炎症反应都可能扩大丘脑和大脑皮层细胞的受损范围[45]。但矛盾的是,在缺血性脑卒中模型鼠使用NLRP3 抑制剂MCC950后,对模型鼠的病变体积以及炎症都没有明显的挽救作用[46]。
尽管NLRP3 炎症小体被认为在中风中发挥较大作用,但NLRP3 炎症小体是否与CCPSP 的发生相关尚未确定。关于CCPSP 的发生机制主要有去抑制理论、中枢敏化和脊髓丘脑束功能改变这三种理论[42]。在去抑制理论中,脊髓丘脑束的损伤破坏了丘脑外侧对丘脑内侧活动的抑制作用,即打破了促进系统和抑制系统间的平衡,抑制通路作用受损,丘脑内侧异常放电导致中枢性疼痛[42]。但是目前没有研究报道NLRP3 与这些机制有直接关联。与NLRP3 相关的炎症和脑损伤可能导致丘脑外侧和脊髓丘脑束功能障碍引起CCPSP,可以成为新的探索方向。
(3)NLRP3 炎症小体与多发性硬化症导致的神经病理性疼痛
多发性硬化症 (multiple sclerosis, MS) 是一种自身免疫性疾病,是由于自身免疫细胞T 细胞激活髓鞘蛋白,以中枢神经系统白质炎性脱髓鞘为主要特点的神经退行性疾病[47],常用的实验模型是自身免疫性脑脊髓炎 (experimental allergic encephalomyelitis, EAE)。
在 EAE 模型中,发病期内后脑 NLRP3 炎症小体表达升高,脑脊液中IL-1β 蛋白表达上调,并且在病灶周围的小胶质细胞中有 GSDMD 和 Caspase-1表达。内质网激活Caspase-1 从而水解GSDMD 后,会介导小胶质细胞发生细胞焦亡。随后,脑组织中驻留的免疫细胞和外周循环白细胞被募集到炎症部位,诱导炎症反应,并导致严重的组织损伤[48]。Nlrp3-/-小鼠的 EAE 模型中临床症状及脱髓鞘的严重程度都有所缓解。采用髓鞘少突胶质细胞糖蛋白诱导的小鼠 EAE 模型中,通过电生理记录发现:DRG 的大、中直径 (≥26 μm) 神经元过度兴奋,NLRP3 炎症小体被长时间激活,神经元和神经胶质细胞中 NLRP3、Caspase-1 和IL-1β 转录水平均有所增加[49]。
在 MS 病人中也有相似发现:脑脊液中 IL-1β基因表达上调,外周血 IL-1β 水平高的病人病情进展更快,也更易复发[47]。对 MS 病人进行基因测序,发现 NLRP3 的遗传负荷增加。但也有文献指出,在MS 病人的血浆中没有检测出 IL-1β,且体外培养外周血单核细胞并激活NLRP3 炎症小体后,IL-1β 的分泌水平与健康人相比并无统计学差异[50]。这些证据说明NLRP3 炎症小体是否能诱导MS 和EAE,有可能取决于机体的免疫强度[51]。
在临床前研究中,EAE 模型有两种亚型,A 型为典型EAE,对 NLRP3 炎症小体有依赖性并且对IFN-β 治疗有反应。大剂量的灭活分枝杆菌加入完全弗氏佐剂后,可以在Nlrp3-/-和Asc-/-小鼠中诱发EAE,此为 B 型。 B 型EAE 可以在没有 NLRP3 炎症小体的情况下发展,并且IFN-β 治疗无效。MS常用治疗药物干扰素 IFN-β-1a/b(倍泰龙/利比)对A 型EAE 有较好的治疗效果,对 B 型 EAE 治疗效果较差。这表明 NLRP3 炎症小体可能是IFN-β 治疗 MS 的潜在关键靶点[51]。已有实验表明给 EAE模型鼠使用 MCC950 可以延缓发作,降低 MS 的严重程度,预防复发并减轻神经性疼痛[52]。这提示NLRP3 炎症小体抑制剂有希望作为新的镇痛剂,用于减轻MS 早期的中枢神经病理性疼痛。
三、小结
本文系统阐述NLRP3 炎症小体参与慢性NP相关研究的最新进展。近年来,越来越多的证据证实NLRP3 炎症小体在各种慢性NP 动物模型的局部组织、外周神经元、脊髓和脑区内被激活。激活的过程以及涉及的机制、上下游因子都被证实与慢性NP 的发作、持续和进展密切相关。使用NLRP3 炎症小体的拮抗剂或通过基因敲除方法进行的研究,进一步证实NLRP3 炎症小体及其下游信号炎症因子参与介导慢性NP 的发生和发展。因此,NLRP3炎症小体被认为是极具潜力的慢性NP 治疗靶点。当然,这一结论还有待更进一步的研究探索加以证实。首先,NLRP3 炎症小体发挥作用的机制较为复杂,其中的镇痛机制还需要进一步的基础及临床研究加以阐明。其次,虽然在动物模型中NLRP3 与慢性疼痛的发生密切相关,但在临床病人中NLRP3是否发挥作用及其生理病理机制仍不明确。最后,NLRP3 的抑制药物目前普遍应用于基础研究,距离步入临床使用还有很长的距离,需要临床阶段的试验评估其在慢性NP 治疗中的安全性和有效性。但许多已经用于临床的药物及中草药中的天然成分也被证实对NLRP3 炎症小体具有抑制效果,对于这部分药物的深入研究有助于在更短的时间内开发出有效的NP 治疗药物。
利益冲突声明:作者声明本文无利益冲突。
