环戊烯与四氧化锇环加成反应的机理研究
2022-10-31刘悦灵陈旺张干兵
刘悦灵,陈旺,张干兵
(湖北大学化学化工学院, 湖北 武汉 430062)
0 引言
环加成反应是合成杂环化合物的常用方法之一.1912年Hoffman首次发现了四氧化锇能够催化氧化烯烃羟基化[1-2]. Sharpless及其合作者[3-4]将四氧化锇应用到烯烃的不对称氧化,合成顺式邻二醇.因四氧化锇催化烯烃的不对称双羟基化反应具有高效率和高选择性,在有机合成中得到了广泛的应用[5-6]. 四氧化锇催化多烯烃的双羟基化反应合成各种杂环化合物的实验研究也见诸报道[7-9].

在上述理论研究中,大多对直链烯烃与四氧化锇的环加成反应机理进行了研究,而对环烯烃与四氧化锇环加成反应的理论研究还很缺乏,本研究运用密度泛函理论方法,研究了环戊烯与四氧化锇的[3+2]环加成以及[2+2]环加成-扩环重排两种机理,以期对四氧化锇与烯烃的环加成反应机理进行补充和完善,为相关实验研究提供理论依据.
1 计算过程
结合相对论赝势,运用M06-2X[21]方法对环戊烯与四氧化锇的[3+2]环加成反应路径以及[2+2]环加成-重排反应路径上的所有驻点(包括反应物、中间体、过渡态以及环加成产物)进行了几何全优化,用频率分析确证优化所得的几何构型是反应路径上的极小值点或过渡态,并计算热力学函数(零点振动能(ZPE)、焓、吉布斯自由能等). 通过内禀反应坐标(IRC)计算以验证所优化的过渡态对前后两个能量极小点的正确连接. 对两个反应路径上的过渡态的优化结构做了自然键轨道(NBO)分析. 锇原子采用SDD相对论赝势基组,非金属原子采用Pople系列的3-ξ基组6-311G**,所有的计算均用G09[22]程序完成.
为了检验所选的方法对本研究体系是否合适,首先采用所选的方法和基组优化了四氧化锇分子的几何结构,并计算了其键解离能.优化得到四氧化锇分子为正四面体结构,Os—O键长d(Os—O)=0.170 0 nm,这与实验值d(Os—O)=0.171 4±0.000 3 nm相符合[23];计算得到四氧化锇的键解离能D(OsO3—O)=67.4 kcal/mol,与实验测值[24]D(OsO3—O)=78.0±14 kcal/mol基本一致.所以本研究采用的计算方法是适合的.
2 结果与讨论
该加成反应的机理如图式(Scheme)1所示,各驻点的优化几何如图1所示,各驻点的相对能量如表1所示,相应的势能曲线如图2所示.
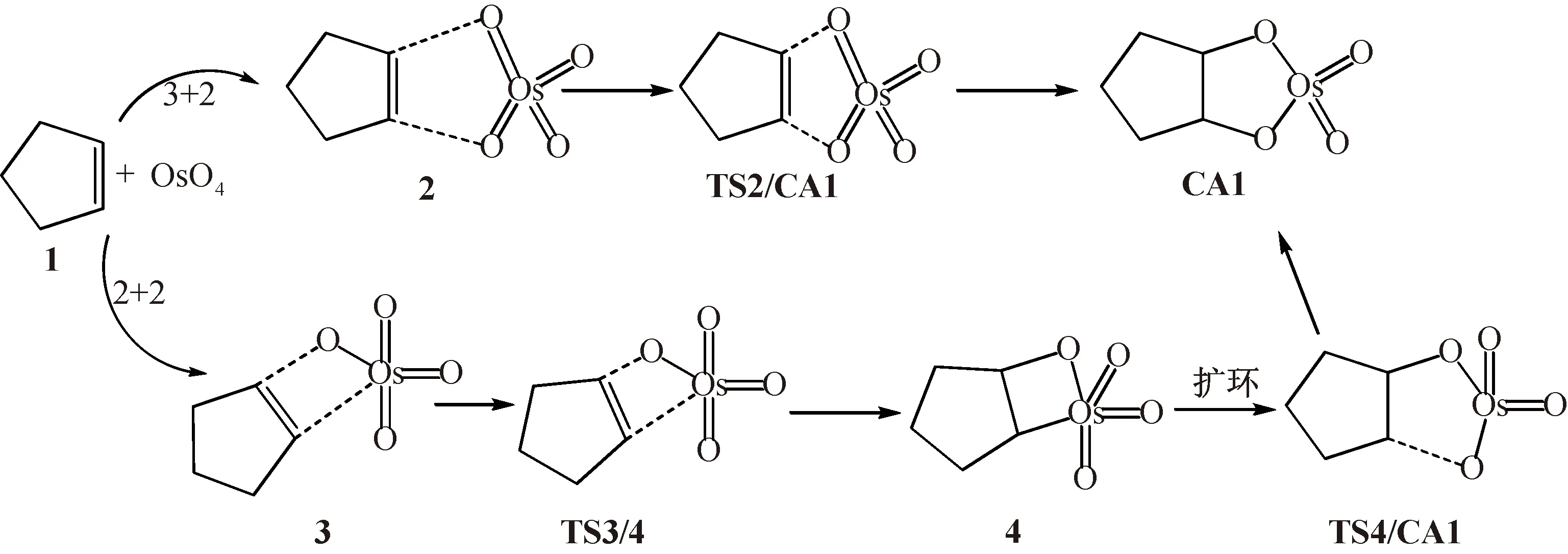
Scheme 1 环戊烯与四氧化锇环加成反应的可能路径
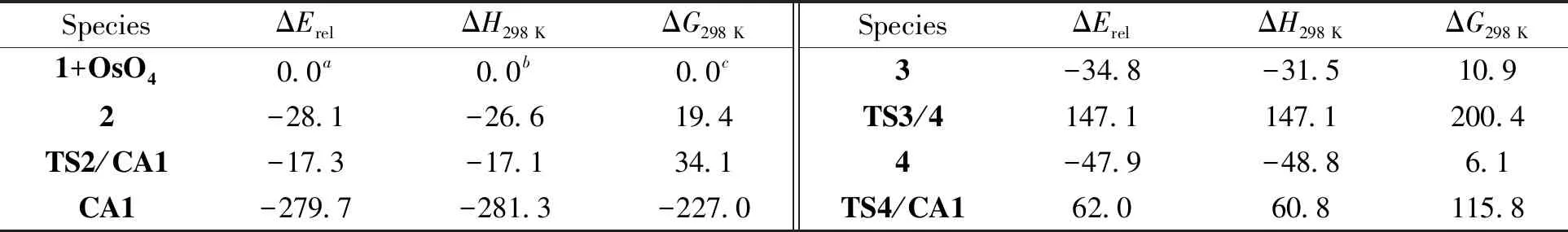
表1 环戊稀与四氧化锇环加成反应的两种可能路径上各驻点的能量(含零点能) kJ/mol
2.1 反应热力学由Scheme1可知,环戊烯(1)与四氧化锇加成不论经历那种路径,最后的氧化产物均为锇酸酯CA1.由表1可知,室温下反应的ΔrG=-227.0 kJ/mol (放热281.3 kJ/mol),是室温下热力学上可能的反应.

图1 M06-2X方法优化环戊烯与四氧化锇环加成反应各驻点的几何构型 (键长单位:0.1 nm,键角单位:°)
2.2 反应历程由Scheme1可知,环戊烯(1)与四氧化锇加成经历协同的[3+2]环加成和[2+2]环加成-扩环两种可能的路径.

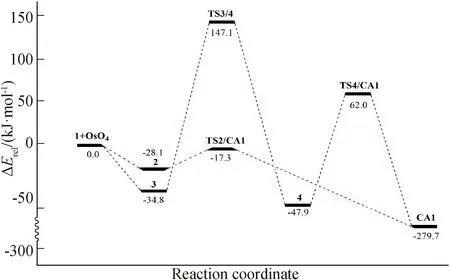
图2 环戊稀与四氧化锇环加成反应两种可能路径上的势能曲线
在扩环重排中,主要涉及C2与O9的耦合成键.计算结果表明,随着C2与O9的不断靠近,C2与Os6不断远离,要形成这样的过渡态结构,四氧化锇部块必须要有较大的几何翻转,扩环过渡态中O9与C2之间的距离变为0.238 9 nm,此值虽与产物中的差别较大,但与反应物中的差别更大.最终,虽然相对于产物而言,此过渡态中扩环涉及到的两个Os—O键和C1—O7键长均与此步的反应物4中的更接近,但由于四氧化锇有着较大的几何变形,该过渡态结构并不具备前期过渡态的特征,所以其活化能比较大,此步能垒为109.9 kJ/mol.
2.2.3 自然键轨道(NBO)分析 为了探讨影响两种加成方式中过渡态能量的影响机制,对所有过渡态都做了自然键轨道(NBO)分析.图3为3+2加成的过渡态TS2/CA1中主要电子供体NBO与受体NBO间的电子转移相互作用以及二级微扰能E(2)(因2+2加成的过渡态和扩环步的过渡态中对应的轨道作用的二级微扰能很小,所以没有列出相互作用的轨道对).由图3可知,在3+2协同加成过渡态中存在着C1—C2间π轨道与O7—Os6的反键π轨道间、O8的孤对电子占据的非键轨道与C1—C2间π*轨道的电子转移相互作用(二级微扰能分别为20.92 kCal/mol和8.66 kCal/mol,体现了他们相互作用的强弱).这些一方面有利于削弱两个原始组块中的双键,促进二者环加成;另一方面,强的电荷转移促进形成较稳定的活化配合物,从而有较低的能垒.这一点由3+2环加成过渡态中从Os向周围配体间的电子转移量1.58e都大于2+2环加成过渡态和扩环过渡态中的相应值(分别为1.44e和1.32e)而得到印证.

图3 过渡态TS2/CA1中存在的主要电子供体NBO与受体NBO间的电子转移相互作用及二微扰能E(2)
3 小结
通过对环戊烯与四氧化锇的环加成反应势能面上各驻点的几何、能量的计算,探讨了有关机理.可得到如下结论:1)整个加成反应的ΔG为-227.0 kJ/mol(放热281.3 kJ/mol),是热力学可能的反应.2)由于[3+2]环加成反应路径中,形成过渡态结构不需要太大的几何形变,其形成时期比[2+2]环加成反应路径的过渡态形成得早,[3+2]环加成所需要翻越的能垒仅为10.8 kJ/mol;[2+2]环加成的能垒比[3+2]环加成要高出171.1 kJ/mol,而经2+2环加成之后的扩环重排过程中,形成过渡态要求四氧化锇部块作较大的几何翻转,需要克服109.9 kJ/mol的位垒,导致2+2环加成-扩环重排路径中需要克服连续两个很高的能垒,在动力学上不利,反应将以[3+2]协同环加成的形式进行.
