新型硒二唑衍生物荧光分子的合成及光电性能研究
2022-10-28蒋振涛林升大张贤慧黄雯孝黄薛龙
江 凡, 蒋振涛, 林升大, 张贤慧, 黄雯孝, 黄薛龙*
(1. 赣南医学院 心脑血管疾病防治教育部重点实验室 药学院,江西 赣州 341000;2. 广东省矿产应用研究所 国土资源部放射性与稀有稀散矿产重点实验室,广东 韶关 512026)
分子内电荷转移(ICT)效应是荧光探针设计采用的最为广泛的作用机理之一。ICT机理的荧光探针结构包括荧光生色团和特征识别基团,其中荧光生色团包括给电子单元(Donor,简称D)和吸电子单元(Acceptor,简称A)。 D-A单元构筑的荧光生色团通过给电子单元和吸电子单元的推拉效应形成电子云的再分布,通过发生光诱导分子内电荷转移为激发态的变化,实现分子吸收光谱的红移和蓝移[1-6]。三苯胺单元具有结构稳定、Stoke位移大的特征,易与强吸电子单元偶联得到高荧光量子效率的共轭分子,是荧光探针设计中一种常见的给电子单元[7-8]。本课题组基于三苯胺(D单元)和苯并噻二唑(A单元)化合物设计合成了一种荧光探针TBBA,可以实现对生物体痕量Hg2+的特异识别。优化荧光分子在NIR区间的覆盖范围,有助于避免光诱导的生物损伤和降低自发荧光,同时还可以提高细胞穿透力,利于荧光探针在生物体检测的实际应用[9]。在保持给电子单元为三苯胺不变的情况下,选择吸电子能力更强的A单元来提高荧光分子的ICT效应,从而达到吸收光谱红移的目的。硒原子相比同主族的硫原子,具有原子半径大、电负性小的特点,因而有望使吸收光谱发生红移[10-14]。
本文基于合成的TBBA,将苯并噻二唑单元换成苯并硒二唑和吡啶硒二唑单元,得到两个新型结构的荧光分子(Z)-2-(5-(4-(7-(4-(二苯胺基)苯基)苯并[c][1,2,5]硒二唑-4-基)苄叉亚甲基)-4-氧代-2-硫代噻唑烷-3-基)乙酸(TBSeBA, Scheme 1)和(Z)-2-(5-(4-(4-(4-(二苯胺基)苯基)-[1,2,5]硒二唑[3,4-c]吡啶-7-基)苄叉亚甲基)-4-氧代-2-硫代噻唑烷-3-基)乙酸(TPSeBA, Scheme 1),以期获得吸收光谱的红移,为近红外荧光探针的设计提供一种新思路。
1 实验部分
1.1 仪器与试剂
Bruker AV-400 MHz型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标);Varioskan LUX-3020型紫外可见光谱仪;Varioskan LUX-3020型荧光分光光度计;FL2000型发射光谱仪;Waters Q-TOF型质谱仪。
4,7-二溴-[1,2,5]硒二唑[3,4-c]吡啶和4,7-二溴苯并[c][1,2,5]硒二唑按文献[10,15]方法合成,4-硼酸三苯胺、4-甲酰基苯硼酸、罗丹宁-3-乙酸、四(三苯基膦)钯、甲苯、四氢呋喃、二苯甲酮。
Scheme 1
1.2 合成
(1) 化合物4的合成
将4-硼酸三苯胺0.8555 g(2.9700 mmol)和4,7-二溴苯并硒二唑1.0100 g(2.9700 mmol)加入到150.00 mL两口瓶中,抽换气3次,氮气保护下加入四(三苯基膦)钯催化剂51.00 mg。用注射器从软管加入干燥甲苯15.00 mol、 10%四丁基氢氧化铵水溶液10.00 mL和干燥四氢呋喃30.00 mL,于120 ℃条件下回流反应24 h。冷却至室温,二氯甲烷萃取,水洗3次,无水硫酸镁干燥,溶剂旋干,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=20/3,V/V)纯化,甲醇重结晶后得橙黄色块状固体4,收率45.2%;1H NMR(CDCl3, 400 MHz,)δ: 7.65(s, 2H), 7.39(d,J=7.5 Hz, 2H), 7.32~7.27(m, 4H), 7.20~7.16(m, 6H), 7.10~7.05(m, 2H);13C NMR(CDCl3,100 MHz)δ: 158.46, 157.25, 148.29, 147.37, 135.28, 132.45, 132.17, 130.55, 130.18, 129.40, 127.60, 125.01, 124.39, 123.46, 122.58, 116.55, 114.94, 35.00, 31.52, 30.14, 29.73; MS(ESI)m/z: Calcd for C24H16N3BrSe[M+]504.97, found 505.97。
(2) 化合物6的合成
将4-(苯并[1,2,5]硒二唑-4-基)-二苯基苯胺0.1652 g(0.3269 mmol)和4-羧基苯硼酸0.0750 g(0.4900 mmol)加入到150.00 mL两口瓶中,抽换气3次,氮气保护下加入四(三苯基膦)钯催化剂0.0566 g(0.0490 mmol)。用注射器从软管加入干燥甲苯15.00 mL、四丁基氢氧化铵10.00 mL和干燥四氢呋喃30.00 mL,于120 ℃条件下加热回流24 h。冷却至室温,二氯甲烷萃取,水洗3次,无水硫酸镁干燥,旋干,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=20/1,V/V)纯化。甲醇重结晶后得红褐色块状固体6,收率68.7%;1H NMR(CDCl3, 400 MHz)δ: 10.04(s, 1H), 7.99(q,J=8.4 Hz, 4H), 7.74(d,J=8.8 Hz, 2H), 7.64(d,J=7.3 Hz, 1H), 7.57(d,J=7.1 Hz, 1H), 7.22(dd,J=15.9 Hz, 8.5 Hz, 5H), 7.17~7.09(m, 6H), 7.01(t,J=7.3 Hz, 1H);13C NMR(CDCl3, 100 MHz)δ: 190.94, 158.57, 147.19, 146.35, 143.10, 134.75, 134.60, 131.82, 130.01, 129.23, 129.05, 128.83, 128.46, 128.36, 126.38, 123.97, 122.39, 121.55; MS(ESI)m/z: Calcd for C31H21N3OSe[M+]531.08, found 537.53。
(3) 化合物TBSeBA的合成
将4-(7-(4-(二苯氨基)苯基)苯并[1,2,5]硒二唑-4-基)苯甲酸0.1340 g(1.0000 mol)和罗丹宁-3-乙酸0.0872 g(1.9600 mol)加入到150.00 mL两口瓶中,抽换气3次,氮气保护下加入乙酸0.2803 g(18.5000 mol)至完全溶解。加入乙酸酐0.0461 g(1.7900 mol)和NH4OAc 0.0433 g(2.2300 mol),于120 ℃条件下反应20 h,再升温至130 ℃反应4 h。冷却至室温,布氏漏斗抽滤,滤渣用冰水冲洗,洗至滤液由黄色变为无色透明。经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=10/1,V/V)纯化、二氯甲烷冲洗,50 ℃真空干燥得红褐色块状固体TBSeBA,收率86.8%;1H NMR(CDCl3, 400 MHz)δ: 7.82(d,J=7.5 Hz, 1H), 7.76~7.69(m, 3H), 7.39(d,J=7.5 Hz, 1H), 7.29(dd,J=8.5 Hz, 7.2 Hz, 6H), 7.22~7.14(m, 9H), 7.10~7.04(m, 3H);13C NMR(CDCl3, 100 MHz)δ: 158.61, 158.47, 148.29, 147.53, 147.37, 135.28, 132.45, 130.56, 130.18, 129.40, 129.35, 127.60, 125.01, 124.87, 123.46, 123.24, 122.58, 114.94; MS(ESI)m/z: Calcd for C36H24N4O3S2Se[M+]704.05, found 702.52。
(4) 化合物4-(7-溴-[1,2,5]硒二唑[3,4-c]吡啶-4-基)-N,N-二苯基苯胺5的合成
将4,7-二溴-[1,2,5]硒代二唑[3,4-c]吡啶1.3674 g(4.0000 mmol)和4-硼酸三苯胺1.1565 g(4.0000 mmol)加入到100.00 mL两口瓶中,抽换气3次,氮气保护下加入四(三苯基磷)钯催化剂0.2311 g(0.2000 mmol)。用注射器从软管加入干燥甲苯32.00 mL、干燥四氢呋喃16.00 mL和四丁基氢氧化铵10.00 mL, 90 ℃条件下加热回流24 h。冷却至室温,水洗3次,二氯甲烷萃取,无水硫酸镁干燥。溶剂旋干,硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=40/1,V/V)纯化。甲醇重结晶得鲜红色固体5,收率39.1%;1H NMR(CDCl3, 400 MHz)δ: 8.62(s, 1H), 8.40(d,J=8.8 Hz, 2H), 7.32(dd,J=8.4 Hz, 7.3 Hz, 4H), 7.22~7.16(m, 5H), 7.16~7.10(m, 3H);13C NMR(CDCl3, 100 MHz)δ: 160.36, 154.99, 153.84, 150.39, 146.85, 145.25, 131.54, 129.52, 129.39, 125.63, 124.16, 121.28, 111.08; MS(ESI): Calcd for C23H15BrN4Se[M+]505.96, found 506.97。
(5) 化合物4-(7-溴-[1,2,5]硒二氮杂[3,4-c]吡啶-4-基)-N,N-二苯基苯胺7的合成
将4-(7-溴-[1,2,5]硒二氮杂[3,4-c]吡啶-4-基)-N,N-二苯基苯胺0.3822 g(0.7534 mmol)和4-甲酰基苯硼酸0.1692 g(1.1301 mmol)加入到100.00 mL两口瓶中,抽换气3次,氮气保护下加入四(三苯基磷)钯催化剂0.2610 g(0.2260 mmol)。用注射器从软管加入干燥甲苯20.00 mL、干燥四氢呋喃10.00 mL和10%四丁基氢氧化铵水溶液8.00 mL,于90 ℃条件下加热回流24 h。冷却至室温,水洗3次,二氯甲烷萃取,无水硫酸镁干燥。溶剂旋干,硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=40/1,V/V)纯化。甲醇重结晶后得暗红色固体7,收率67.6%;1H NMR(CDCl3, 400 MHz)δ: 10.12(s, 1H), 8.63(s, 1H), 8.49~8.43 (m, 2H), 8.14(d,J=8.4 Hz, 2H), 8.09~8.03(m, 2H), 7.35~7.30(m, 4H), 7.24~7.18(m, 6H), 7.15~7.10(m, 2H);13C NMR(CDCl3, 100 MHz)δ: 191.89, 160.95, 155.45, 150.39, 146.87, 141.40, 135.91, 131.67, 130.05, 130.00, 129.52, 126.47, 125.64, 124.16, 121.31; MS(ESI)m/z: Calcd for C30H20N4OSe[M+]532.08, found 533.08。
(6) 化合物TPSeBA的合成
将4-(7-溴-[1,2,5]硒二氮杂[3,4-c]吡啶-4-基)-N,N-二苯基苯胺0.2240 g(0.4206 mmol)、罗丹宁-3-乙酸0.1576 g(0.8243 mmol)和醋酸铵0.0730 g(0.9479 mmol)加入10.00 mL两口瓶中,抽换气3次,氮气保护下加入乙酸酐0.1548 g(1.5058 mmol)和冰乙酸1.8690 g(31.1244 mmol),油浴下加热至120 ℃反应20 h,再升温至130 ℃反应4 h。冷却至室温,布氏漏斗抽滤,滤渣用冰水冲洗,洗至滤液由黄色变为无色透明,继续用二氯甲烷冲洗。真空干燥得紫黑色块状固体TPSeBA,收率66.9%;1H NMR(DMSO-d6, 400 MHz)δ: 13.50(s, 1H), 8.71(s, 1H), 8.54~8.49(m, 2H), 8.25(d,J=8.2 Hz, 2H), 7.99(s, 1H), 7.85(d,J=8.3 Hz, 2H), 7.43~7.36(t,J=8.0 Hz, 4H), 7.17(dd,J=7.7 Hz, 4.2 Hz, 6H), 7.09~7.05(m, 2H), 4.77(s, 2H);13C NMR(DMSO-d6, 100 MHz)δ: 193.58, 166.88, 153.37, 146.90, 142.48, 138.32, 133.92, 132.09, 131.43, 130.74, 130.29, 126.20, 125.72, 124.75, 120.95, 45.53, 21.54; MS(ESI)m/z: Calcd for C35H23N5O3S2Se[M+]705.04, found 706.04。
2 结果与讨论
2.1 合成
荧光分子合成路线如Scheme 1所示。4-(二苯基氨基)苯硼酸与4,7-二溴苯并[c][1,2,5]硒二唑在钯催化下通过Suzuki偶联得到化合物4,化合物4与4-甲酰基苯硼酸偶联得到化合物6,化合物6与识别基团罗丹宁-3-乙酸加成得到目标分子TBSeBA。采取类似的合成路线,用4,7-二溴苯并[c][1,2,5]硒二唑替换4,7-二溴-[1,2,5]硒代二唑[3,4-c]吡啶可以得到目标分子TPSeBA。
合成路线中的所有单体和目标产物均通过核磁共振氢谱碳谱表征,TBSeBA为红褐色块状固体,TPSeBA为紫黑色块状固体。其中甲苯和四氢呋喃处理方法为:甲苯用浓硫酸洗涤3次,再用碳酸氢钠水溶液和去离子水分别洗涤3次,经无水硫酸镁干燥过夜后待用;四氢呋喃在氮气环境下用金属钠干燥,回流24 h后加入二苯甲酮变成蓝色后待用。
2.2 表征
(1) 光学性能
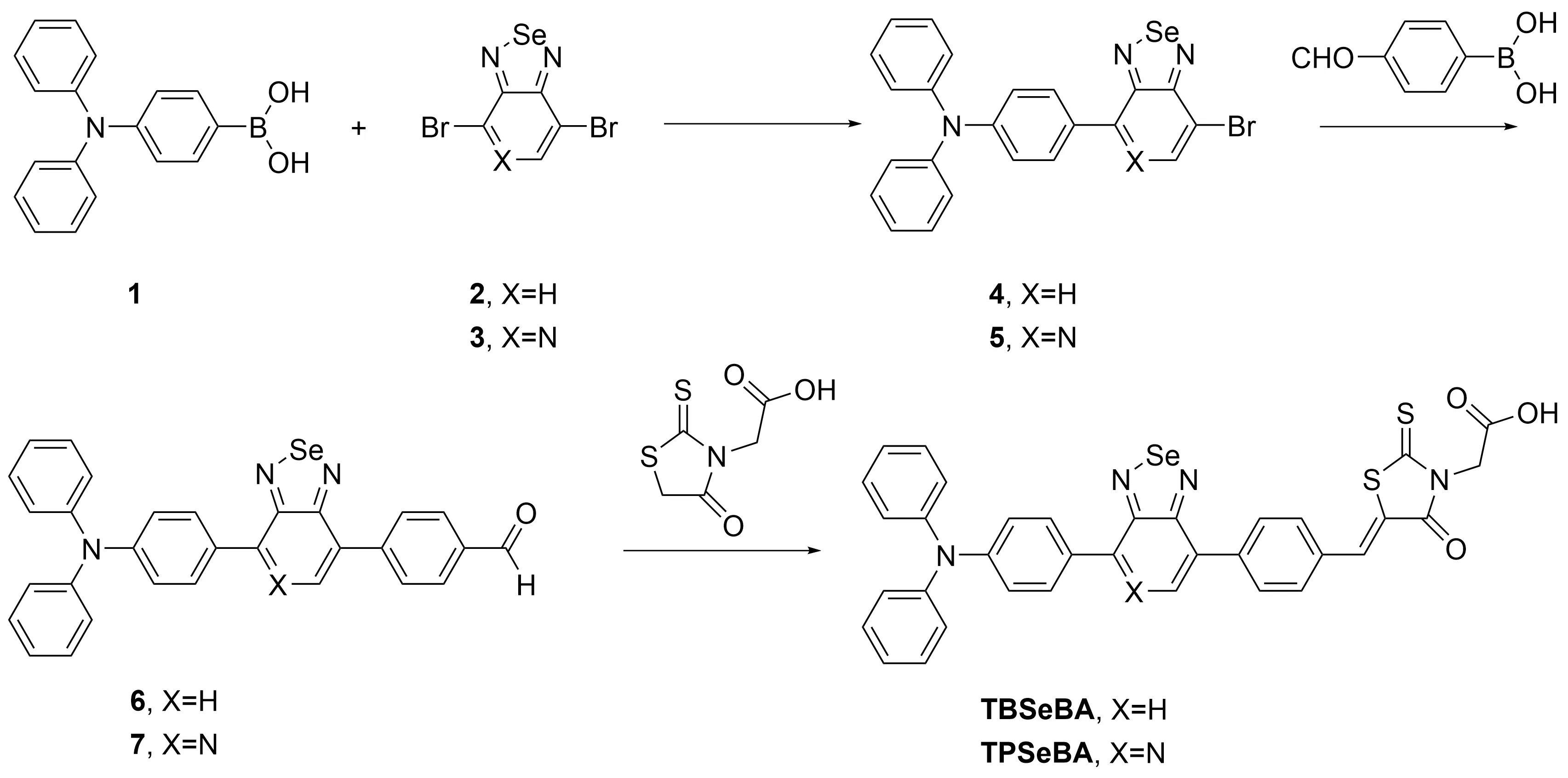
图1为TPSeBA和TBSeBA在DMSO溶液中的紫外-可见吸收光谱。TPSeBA和TBSeBA在350~450 nm和450~600 nm处均呈现一个吸收带,其中350~450 nm吸收带属于三苯胺等共轭单元所产生的π-π*跃迁吸收,450~600 nm的吸收带归属于给电子单元三苯胺与吸电子单元苯并硒二唑/吡啶硒二唑之间形成的分子内电荷转移峰(ICT)。TBSeBA在溶液中的吸收峰为465 nm,而TPSeBA的吸收峰红移至510 nm,该现象归因于吡啶硒二唑比苯并硒二唑具有更强的吸电子能力。图2为TPSeBA和TBSeBA的发射光谱。TPSeBA最大荧光发射峰为744 nm,TBSeBA最大荧光发射峰为646 nm,两个峰均属于红光区域,其中TPSeBA相较于前期合成的TBBA,其最大荧光发射峰有发生近70 nm红移。

λ/nm图1 化合物的UV-Vis谱图Figure 1 UV-Vis spectra of compounds
λ/nm图2 化合物的FL谱图Figure 2 FL spectra of compounds
(2) 电化学性能
通过循环伏安法测定TPSeBA和TBSeBA的电化学性质。在CHI 660A电化学工作站中,以六氟磷酸四正丁基铵(Bu4NPF6)-DMSO为溶液,采用标准的三电极电化学电池来进行测定(饱和甘汞电极为参比电极,铂碳电极为工作电极,铂丝为辅助电极,以二茂铁(Fc+/Fc)为内标)。氧化过程中TPSeBA的起始电压为1.50 eV,而TBSeBA的起始电压为1.51 eV,这主要是由于吸电子单元结构的差异引起的,以相同方法测得TPSeBA还原电位为-0.66 eV,TBSeBA还原电位为-0.70 eV(图3)。
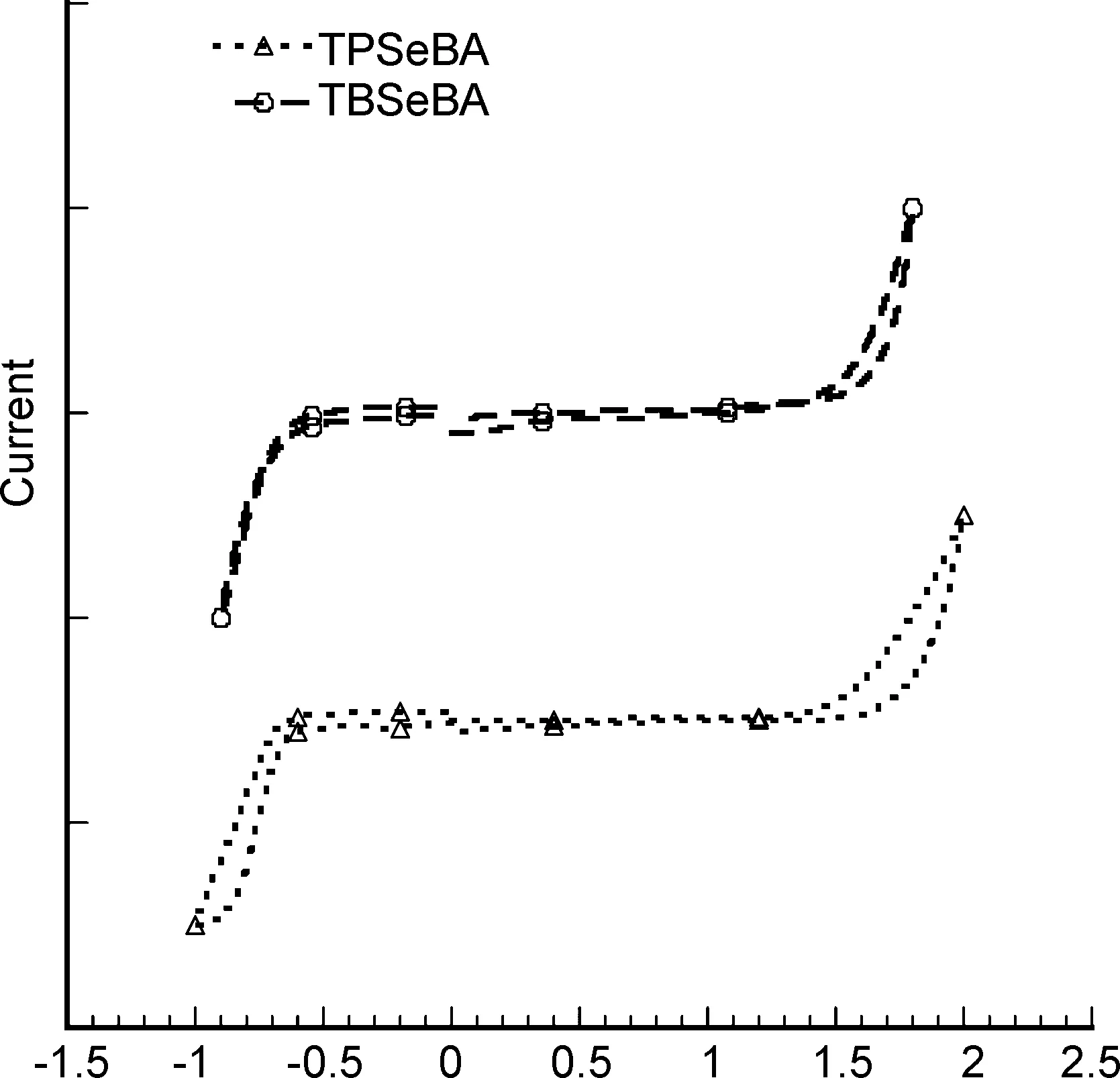
Potential vs. Fc/Fc+(V)图3 化合物的CV曲线Figure 3 CV curves of compounds

表1 荧光电化学与吸收性质Table 1 Optical and electrochemical properties of compounds
二茂铁(Fc+/Fc)的真空能级为4.8 eV,测得其起始电位为0.52 eV。最高占有分子轨道能级(HOMO)可根据公式计算得出:EHOMO=-(Eox+4.28) eV,最低未占有分子轨道能级(LUMO)可根据公式ELUMO=-(Ered+4.28) eV。根据上述公式,TPSeBA和TBSeBA的HOMO能级分别为-5.78 eV和-5.79 eV, LUMO能级分别为-3.62 eV和-3.58 eV(表1)。
以三苯胺为给电子单元,苯并硒二唑和吡啶硒二唑为吸电子单元,罗丹宁为识别基团,设计合成了两种新型荧光分子TBSeBA和TPSeBA。在DMSO溶液中,TBSeBA和TPSeBA的吸收峰分别位于465 nm 和 520 nm。通过循环伏安法测得TBSeBA的HOMO和LUMO能级分别为-5.00 eV和-2.82 eV;TPSeBA的HOMO和LUMO能级分别为-5.17 eV和-2.77 eV。本文采用引入硒原子的策略对荧光分子进行光谱和能级的有效调控,为荧光探针类分子的光谱调控提供一种新思路。
