一锅法合成2-羟基-9-芴酮类化合物
2022-10-28朱启萌姜雅坤武超芊毛亚兰范晓语王程宇
朱启萌, 姜雅坤, 武超芊, 毛亚兰, 范晓语, 王程宇
(临沂大学 化学化工学院,山东 临沂 276000)
芴酮骨架大量存在于具有生理活性的天然产物或药物分子中,呈现出各种不同的药效价值[1-2]。1970年,Krueger等[3]发现化合物Tiorone dihydrochloride在老鼠体内具有抗病毒药理作用,之后研究者们陆续发现该化合物还具有增强自然杀伤细胞的吞噬活性[4]、解热[5]、抗纤维化[6]、消炎[7]以及干扰素诱生[8]等药理活性,该药效作用部分归因于芴酮骨架的平面结构修饰[9]。
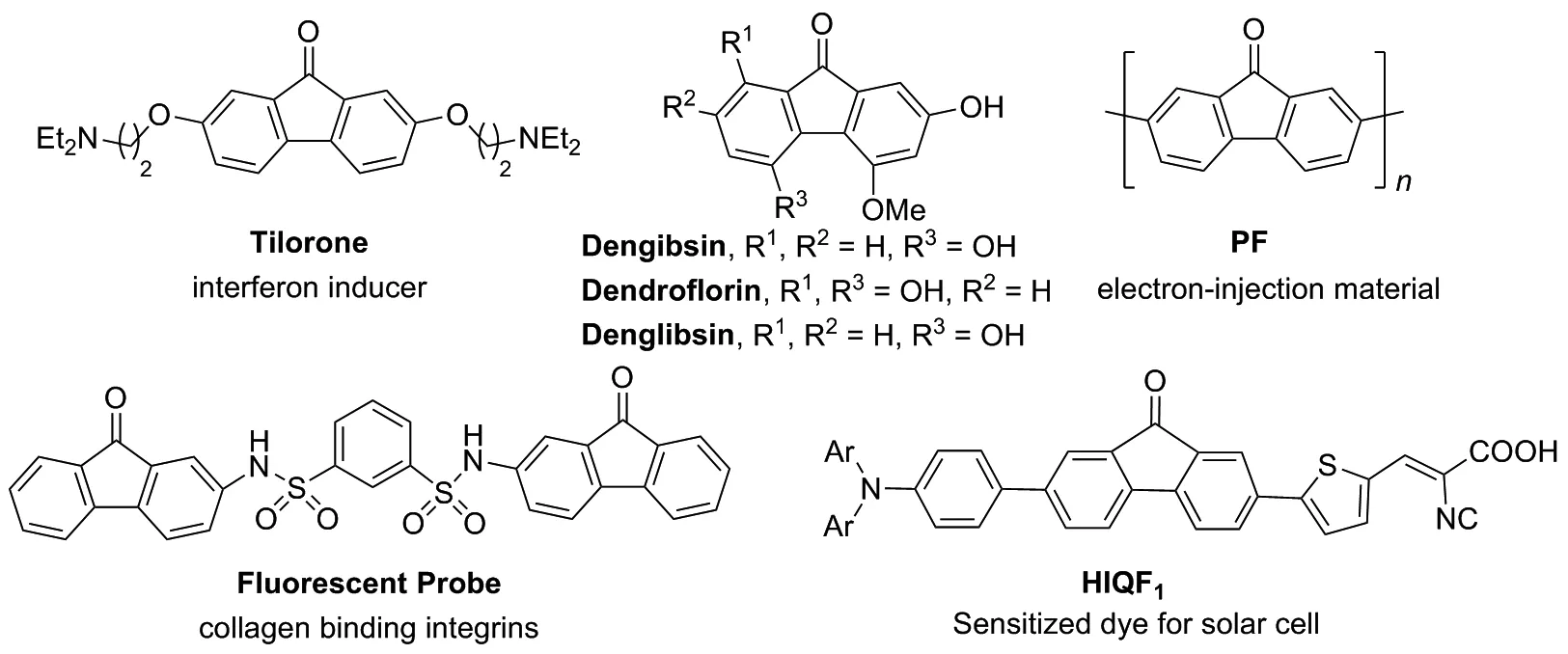
Chart 1

Scheme 1
此外,芴酮类化合物由于其独特的光电性能,也常作为可调控的合成子,应用于有机半导体光电聚合材料领域[10]。例如,聚合物PF可以作为电子注射材料[11],HIQF1可以作为太阳能电池敏化染料[12]等(Chart 1)。由于芴酮类化合物显著的药理活性和独特的光电性能,其构建方法引起了有机化学家们的广泛关注。
芴酮骨架的合成策略大致可以分为两类:分子内反应和分子间反应。分子内反应常用的反应底物包括邻羰基联苯类化合物[13-14]、二苯甲酮衍生物[15-17]和三炔类化合物[18]等,通过傅克酰基化、氧化环化、过渡金属催化分子内C—H键活化和[4+2]环加成等反应模式均可构建芴酮分子。2006年,Langer等[13]报道了通过多步反应合成邻甲酸酯联苯类化合物,之后在酸性条件下发生分子内傅克酰基化反应得到芴酮衍生物;2013年,Glorius课题组[14]报道了邻甲酰基联苯类化合物在过氧化物存在条件下,底物中甲酰基中的C—H键均裂产生酰基自由基,进而分子内自由基环化得到芴酮骨架;2018年,Hoye等[18]报道了邻共轭双炔芳基炔酮底物在酸性条件下发生分子内[4+2]反应得到芴酮类化合物。然而,这些方法往往需要多步合成反应底物,且反应体系多涉及强酸、过氧化物和较高温度等不利因素。
分子间反应常见的反应模式包括:(1) 2-卤联苯类化合物与CO[19]、甲酸苯酯[20]等,过渡金属催化插羰环化反应。2002年,Larock课题组[19]报道了2-卤联苯类化合物在Pd催化下氧化加成得到C-PdX物种,进而与CO发生插羰环化反应得到芴酮衍生物。(2)邻卤苯甲醛与芳基硼酸[21]、卤代芳基硼酸[22]以及苯炔前体[23]等,偶联环化反应。2010年,Ray等[22]报道了一种在过渡金属催化下邻溴苯甲醛与邻溴芳基硼酸经分子间偶联环化反应构建芴酮类化合物。该反应首先发生Suzuki偶联反应得到邻(2-卤苯基)苯甲醛类化合物,进而经过氧化加成、分子内插羰、还原消除得到相应产物。(3)含导向基团的芳基化合物与卤代芳烃[24]、芳基硼酸[25]等,分子间C—H键活化构建芴酮骨架,导向基团可以为肟醚、酰胺、胺、氰基、羧酸和醛基等(Scheme 1)。2010年,Shi课题组[25]报道了芳基肟醚与芳基硼酸在过渡金属催化下经分子间反应构建芴酮衍生物。该反应以肟醚为导向基团,在Pd催化下形成环Pd中间体,进而与芳基硼酸金属交换,还原消除得到邻苯基芳基肟醚中间体,再经进一步环化水解得到相应产物。2013年,Hsieh课题组[26]报道了邻氰基联苯类化合物与卤苯分子间串联环化反应合成芴酮类化合物。该反应以氰基作为导向基团,经C—H键活化、氧化加成和还原消除得到邻氰基联苯类化合物,该化合物再经进一步环化,水解得到相应产物。然而这些方法往往涉及贵金属催化和复杂配体使用,且部分反应的收率不高。因此,发展廉价、温和高效的芴酮骨架构建方法十分必要。

Scheme 2

Scheme 3
本文设计并合成了不同取代类型的邻呋喃芳基炔酮底物(1),并利用其分子内呋喃环与炔键的D-A反应,在酸性催化条件下环氧开环并发生芳构化,一锅法合成2-羟基-9-芴酮衍生物(2a~2e, Scheme 2),收率89%~96%,化合物2a~2e结构经1H NMR,13C NMR和HR-MS(ESI)表征。最后,将合成的产物2e作为底物按照文献方法[27]完成了化合物Tilorone的合成(Scheme 3)。
1 实验部分
1.1 仪器与试剂
Bruker Avance III 400 MHz型核磁共振仪;Agilent 6540 Q-TOF型高分辨质谱仪。
不同取代类型的邻溴苯甲醛、三丁基呋喃锡、氟化钾、不同类型的末端炔、正丁基锂、邻碘酰苯甲酸(IBX)、甲苯、一水合对甲苯磺酸等。所有试剂均为分析纯。
1.2 合成
(1) 邻呋喃芳基炔酮底物(1a)的合成
在氮气氛围下向反应管中依次加入邻溴苯甲醛1.16 mL(10.00 mmol)、甲苯(20.00 mL)、三丁基呋喃锡3.50 mL(11.00 mmol)、 Pd(dba)2115.00 mg(0.10 mmol)和PPh3210.00 mg(0.80 mmol),封管110 ℃条件下反应3~5 h(TLC检测)。待反应原料消失后,加入饱和氟化钾溶液淬灭反应,有大量沉淀产生。反应液经硅藻土过滤,滤液用乙酸乙酯(3×20.00 mL)萃取,有机相用饱和食盐水洗涤(30.00 mL),无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯 ∶石油醚=1 ∶25,V∶V)纯化得化合物邻呋喃苯甲醛化合物1.49 g,收率86%。
氮气氛围下,在干燥的Schlenk反应管中依次加入苯乙炔1.52 mL(13.79 mmol)和无水THF(15.00 mL)。将反应管置于-78 ℃条件下,加入正丁基锂5.20 mL(12.93 mmol)并在该条件下保持30 min。将上一步合成的邻呋喃苯甲醛化合物1.48 g(8.62 mmol)溶于无水THF(5.00 mL)中,并注入反应体系。之后将反应温度降至室温并反应1~3 h(TLC检测)。反应结束后,加水(30.00 mL)淬灭,乙酸乙酯(3×20.00 mL)萃取,有机相用饱和食盐水洗涤(30.00 mL),无水硫酸钠干燥,减压浓缩得到邻呋喃芳基炔醇粗产品,并直接用于下一步反应。
空气氛围下,用15.00 mL二甲亚砜分2~3次将上一步产品转移至100.00 mL圆底烧瓶中。加入邻碘酰苯甲酸(IBX) 2.90 g(10.34 mmol, 1.20 eq),室温过夜反应(TLC检测)。反应结束后加水淬灭,产生大量白色沉淀。反应液经硅藻土过滤,滤液用乙酸乙酯(3×20.00 mL)萃取,有机相用饱和食盐水洗涤(30.00 mL),无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯 ∶石油醚=1 ∶10,V∶V)纯化得邻呋喃芳基炔酮化合物1a2.00 g,收率85%,棕色固体。用类似方法合成反应底物1b~1e,收率83%~95%。
(2) 2-羟基-9-芴酮衍生物(2)的合成
空气氛围下向封管中依次加入反应底物1a82.00 mg(0.30 mmol)、甲苯(3.00 mL)和一水合对甲苯磺酸5.70 mg(0.03 mmol),并在封管100 ℃油浴条件下反应8~17 h(TLC检测)。反应结束后,加入10.00 mL乙酸乙酯稀释并转移至100.00 mL茄形瓶,减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯 ∶石油醚=1 ∶ 4,V∶V)纯化得2-羟基-9-芴酮化合物77.60 mg,收率95%,暗红色固体。用类似方法合成2a~2f。
2a:暗红色固体,收率95%, m.p.194~196 ℃;1H NMRδ: 5.32(s, 1H), 7.07(d,J=8.0 Hz, 1H), 7.17(d,J=3.2 Hz, 1H), 7.37~7.43(m, 5H), 7.47~7.54(m, 5H);13C NMRδ: 119.526, 120.34, 120.84, 124.06, 127.61, 127.92, 128.98, 129.15, 129.62, 131.21, 131.60, 134.14, 134.63, 137.45, 144.05, 154.15, 192.91; HR-MS(ESI)m/z: Calcd for C19H12O2{[M+H]+}273.0916, found 273.0920。
2b:深红色固体,收率94%, m.p.206~207 ℃;1H NMRδ: 7.09(d,J=8.0 Hz, 1H), 7.27~7.31(m, 3H), 7.36~7.39(m, 4H), 7.66(d,J=8.0 Hz, 1H), 7.80(d,J=1.6 Hz, 1H), 10.03(s, 1H);13C NMRδ: 120.11, 120.44, 121.77, 124.92, 127.28, 127.43, 128.92, 130.03, 131.56, 132.02, 133.09, 133.97, 139.80, 145.87, 156.86, 190.99; HR-MS(ESI)m/z: Calcd for C19H11ClO2{[M+H]+}307.0520, found 307.0512。
2c:红色固体,收率90%, m.p.210~213 ℃;1H NMRδ: 6.97~7.02(m, 1H), 7.09(d,J=8.0 Hz, 1H), 7.30~7.34(m, 2H), 7.35~7.41(m, 3H), 7.43(dd,J1=8.2 Hz,J2=5.6 Hz, 1H), 7.56(dd,J1=9.0 Hz,J2=2.0 Hz, 1H), 7.62(d,J=8.0 Hz, 1H), 10.02(s, 1H);13C NMRδ: 107.73(d,J=24.6 Hz), 114.01(d,J=23.4 Hz), 120.27, 121.66, 125.84(d,J=10.4 Hz), 127.22, 127.27, 128.78, 129.82, 130.06, 131.85, 133.12, 133.76, 147.12(J=10.6 Hz), 156.84, 166.85(d,J=250 Hz), 190.65; HR-MS(ESI)m/z: Calcd for C19H11FO2{[M+H]+}291.0816, found 291.0808。
2d:红色固体,收率89%, m.p.264~265 ℃;1H NMRδ: 3.77(s, 3H), 3.92(s, 3H), 6.92(s, 1H), 6.95(d,J=8.0 Hz, 1H), 7.24(s, 1H), 7.28~7.40(m, 6H), 9.61(s, 1H);13C NMRδ: 55.67, 55.96, 103.43, 106.75, 119.11, 119.63, 125.66, 126.86, 126.98, 128.57, 130.01, 131.78, 133.41, 134.90, 139.02, 148.28, 154.58, 155.36, 191.32; HR-MS(ESI)m/z: Calcd for C21H16O4{[M+H]+}333.1121, found 333.1117。
2e:红色固体,收率96%, m.p.269~271 ℃;1H NMRδ: 8.77(s, 2H), 7.40(d,J=8.0 Hz, 2H), 7.01(d,J=2.4 Hz, 2H), 6.95(dd,J1=8.0 Hz,J2=2.4 Hz, 2H);13C NMRδ: 193.96, 158.62, 137.50, 136.83, 121.76, 121.73, 111.97; HR-MS(ESI)m/z: Calcd for C13H8O3{[M+H]+}213.0552, found 213.0556。
(3)Tilorone的合成
按文献报道的方法[27],以2,7-二羟基芴酮(2e)为反应原料,完成了化合物Tilorone的合成。在反应管中依次加入2-氯-N,N-二乙基二胺盐酸盐(95.50 mg, 0.56 mmol, 3.70 eq)、 2,7-二羟基芴酮(2e)(31.80 mg, 0.15 mmol, 1.00 eq)、 KOH(71.50 mg, 1.28 mmol, 8.50 eq)、甲苯(1.20 mL)和H2O(0.30 mL)。将反应体系置于110 ℃油浴条件下回流加热24 h(TLC检测)。反应结束后,加水(30.00 mL)淬灭反应,乙醚(3×15.00 mL)萃取,合并有机相并用无水硫酸钠干燥。减压浓缩,残余物经硅胶柱层析(硅胶预先用5% NEt3二氯甲烷溶液浸泡)(洗脱剂:二氯甲烷 ∶甲醇=20 ∶1,V∶V)纯化得化合物Tilorone40.00 mg,收率65%,橙色固体。1H NMRδ: 1.07(t,J=7.1 Hz, 12H), 2.64(q,J=7.1 Hz, 8H), 2.87(t,J=6.1 Hz, 4H), 4.06(t,J=6.1 Hz, 4H), 6.94(dd,J1=8.1 Hz,J2=2.5 Hz, 2H), 7.15(d,J=2.4 Hz, 2H), 7.25~7.28(m, 2H);13C NMRδ: 12.01, 48.00, 51.73, 67.23, 110.49, 120.62, 120.91, 136.06, 137.60, 159.40, 193.94; HR-MS(ESI)m/z: Calcd for C25H35N2O3{[M+H]+}411.2648, found 411.2698。
2 结果与讨论
2.1 反应条件优化
以10% TsOH·H2O为催化剂,DCM为反应溶剂,在封管100 ℃条件下反应15 h。TLC检测反应结束,直接旋干过柱,以86%的收率得到2-羟基-9-芴酮化合物2a,产物结构经核磁共振氢谱、碳谱以及X-ray单晶衍射确证(CCDC: 2144829)[18](图1)。之后在反应催化剂为10% TsOH·H2O、反应温度为100 ℃的条件下,考察不同极性大小溶剂如DMF、 1,4-dioxane和toluene等对反应效率的影响(表1, Entry 2~4),结果表明,toluene作为反应溶剂时的效果最好(Entry 4)。随后进一步考察了在反应溶剂为toluene,反应温度为100 ℃的条件下,不同类型的路易斯催化剂如FeCl3、 ZnCl2、 AlCl3和BF3·Et2O等对反应效率的影响(表1, Entry 5~8)。由表1可以看出,这些常见的路易斯酸催化剂均能较好地催化反应并能顺利得到目标产物,收率70%~93%。此外,在不加催化剂的情况下反应19 h,以60%收率得到产物(Entry 9)。而当降低反应温度(Entry 10)或减少TsOH·H2O催化剂用量(Entry 11)时,产物收率均有不同程度下降。因此,该反应的最佳反应条件为:10% TsOH·H2O为反应催化剂,toluene为溶剂,反应温度为100 ℃。

图1 化合物2a的单晶结构Figrue 1 Single crystal structure of compound 2a
2.2 反应底物拓展
在最佳反应条件下,通过改变邻呋喃芳基炔酮底物1中R1、 R2取代基进行反应普适性考察。研究结果表明,母环上取代基R1无论是吸电子基(4-Cl、 4-F)还是供电子基(4,5-二甲氧基、5-OH),均能以较优收率得到2-羟基-9-芴酮产物(89%~94%)。 R2无论是芳基炔还是末端炔,同样也能顺利得到相应产物(95%~96%)。因此,该反应的普适性良好。

表1 反应条件优化

Scheme 4
2.3 反应机理
该反应的可能反应机理如Scheme 4所示。反应底物1a在催化剂及加热条件下,发生分子内D-A反应得到氧杂桥环化合物C,化合物C进而在酸性催化条件下发生环氧开环、芳构化过程得到2-羟基-9-芴酮化合物2a。
本文发展了一种合成2-羟基-9-芴酮类化合物的方法,以邻呋喃芳基炔酮为反应底物,利用其分子内D-A反应、环氧开环和芳构化反应,一锅法合成了5种新型的2-羟基-9-芴酮衍生物,收率89%~96%。该方法原料易得,催化体系简单,低成本,反应条件相对温和,且产物中2-位含羟基,便于进一步反应获得衍生物。
