ATP1A2基因突变所致的偏瘫型偏头痛2例报道
2022-10-21丁思奇黄啸君詹飞霞田沃土
丁思奇,黄啸君,詹飞霞,田沃土,曹 立
1. 上海交通大学医学院附属第六人民医院神经内科,上海 200233;2. 安徽医科大学附属宿州市立医院神经内科,宿州234000;3.上海交通大学医学院附属瑞金医院神经内科,神经病学研究所,上海 200025
偏头痛是常见的原发性神经血管疾病,约1/3 患者头痛伴有先兆症状[1]。偏瘫型偏头痛是一种具有临床和遗传异质性的有先兆偏头痛,人群发病率约为1∶20 000[2]。根据一级或二级亲属是否符合偏瘫型偏头痛诊断标准,可将疾病分为家族性偏瘫型偏头痛和散发性偏瘫型偏头痛[3]。遗传学研究发现,家族性偏瘫型偏头痛主要与编码离子通道和转运蛋白的4 种 基 因 (CACNA1A、ATP1A2、SCN1A和PRRT2)相关,CACNA1A和ATP1A2基因突变也可引起散发性偏瘫型偏头痛[4]。ATP1A2相关的偏瘫型偏头痛病例大多数在欧洲和美国报道[5],亚洲病例报道少见。本文收集2 例ATP1A2基因突变相关偏瘫型偏头痛患者的临床资料,结合文献复习进行分析和讨论。
1 临床资料
1.1 基本资料
2 例患者分别于2018 年3 月和2021 年9 月就诊于上海交通大学医学院附属瑞金医院神经内科和上海交通大学医学院附属第六人民医院神经内科,依据临床表型,参照《国际头痛疾病分类(第三版)》诊断标准[1],确诊为偏瘫型偏头痛。
病例1,女性,17 岁。因“反复发作性头痛伴肢体无力13 年余”就诊。患者13 年前(4 岁)开始出现发作性头痛,头痛前有视觉先兆,表现为视野范围出现雪花状模糊,由点状到片状逐渐扩大,持续约30 min,之后出现一侧肢体无力,偏侧肢体完全无法活动。伴对侧头痛,疼痛程度逐渐加重,可伴有恶心、呕吐,呕吐之后头痛可稍缓解,头痛持续12~24 h。偏侧肢体无力症状一般持续1 h 后好转,但在头痛剧烈时可持续4~5 h。头痛缓解后有乏力、头昏不适,可持续1 d 左右。上述症状发作频率约为1 次/月,与月经周期无关;但在感冒、疲劳情况下容易诱发,可达2 次/月。右侧头痛发作频率较左侧高(3∶1),无双侧发作情况。曾服用布洛芬,但效果不佳。有抑郁症病史6 个月,不规律口服盐酸舍曲林,就诊时情绪稳定。父亲和祖母有头痛病史,临床表现与先证者相似,偏头痛前有视觉先兆,伴有一侧肢体无力及麻木感,持续约10 min 后明显缓解,随后出现单侧搏动性头痛,并伴有恶心、呕吐。在呼吸道感染、疲劳时容易诱发,症状发作频率为每3~4 年1 次。患者父母否认近亲婚配。经神经系统检查,患者神志清楚,言语流利,12 对颅神经检查未见异常,四肢肌力、肌张力正常,共济运动正常。头颅CT、磁共振成像、脑电图未见异常。
病例2,女性,14 岁。因“反复半边肢体麻木乏力伴头痛5 年”就诊。患者5 年前(9 岁)起反复出现偏侧肢体麻木、乏力,症状多从下肢起始,逐渐发展至上肢,并累及面部、舌头,影响言语,同时伴眼前闪光点及畏光。肢体麻木无力持续30 min后开始出现头痛,多以右侧顶枕部为主,位置固定,持续4~5 h,呈搏动性,偶有恶心、呕吐,面色苍白,布洛芬、加巴喷丁、普瑞巴林等药物不能缓解症状,睡眠后可好转,发作后全身乏力。肢体麻木乏力症状以单侧为主,左右交替,无双侧同时发作,平均发作频率为1~2 次/年。父母无相似临床症状及其他神经系统疾病,否认近亲婚配。经神经系统检查,患者神志清楚,言语流利,12 对颅神经检查未见异常,四肢肌力、肌张力正常,共济运动正常。血、尿、便常规检查未见异常。凝血功能、肝肾功能、血脂、电解质、脑脊液生化抗体免疫球蛋白、免疫四项、头颅磁共振、脑电图检查均未见异常。
1.2 基因检测
抽取2 例先证者肘静脉外周血,进行全外显子测序(whole exome sequencing,WES),结果提示2 例患者均存在ATP1A2基因错义突变, 分别为c.G2284A/p.G762S(图1A)和c.C3022T/p.R1008W(图1B)。其中变异c.G2284A/p.G762S既往曾有报道,表型为单纯家族性偏瘫型偏头痛[6]。病例1患者父亲和祖母均有类似偏瘫型偏头痛病史,根据临床表现可诊断为家族性偏瘫型偏头痛;但由于家系成员未获得相应血样,故未能进一步行共分离检测。c.C3022T/p.R1008W 既往无单独致偏瘫型偏头痛的相关文献报道。根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics,ACMG) 指 南[7],变 异c.G2284A/p.G762S 被 评 为“ 可 能 致 病”(PS1+PM1+PM2+PP3+PP4), 变 异c.C3022T/p.R1008W 变异性质为“意义不明确”(PM2+PP3+PP4)。
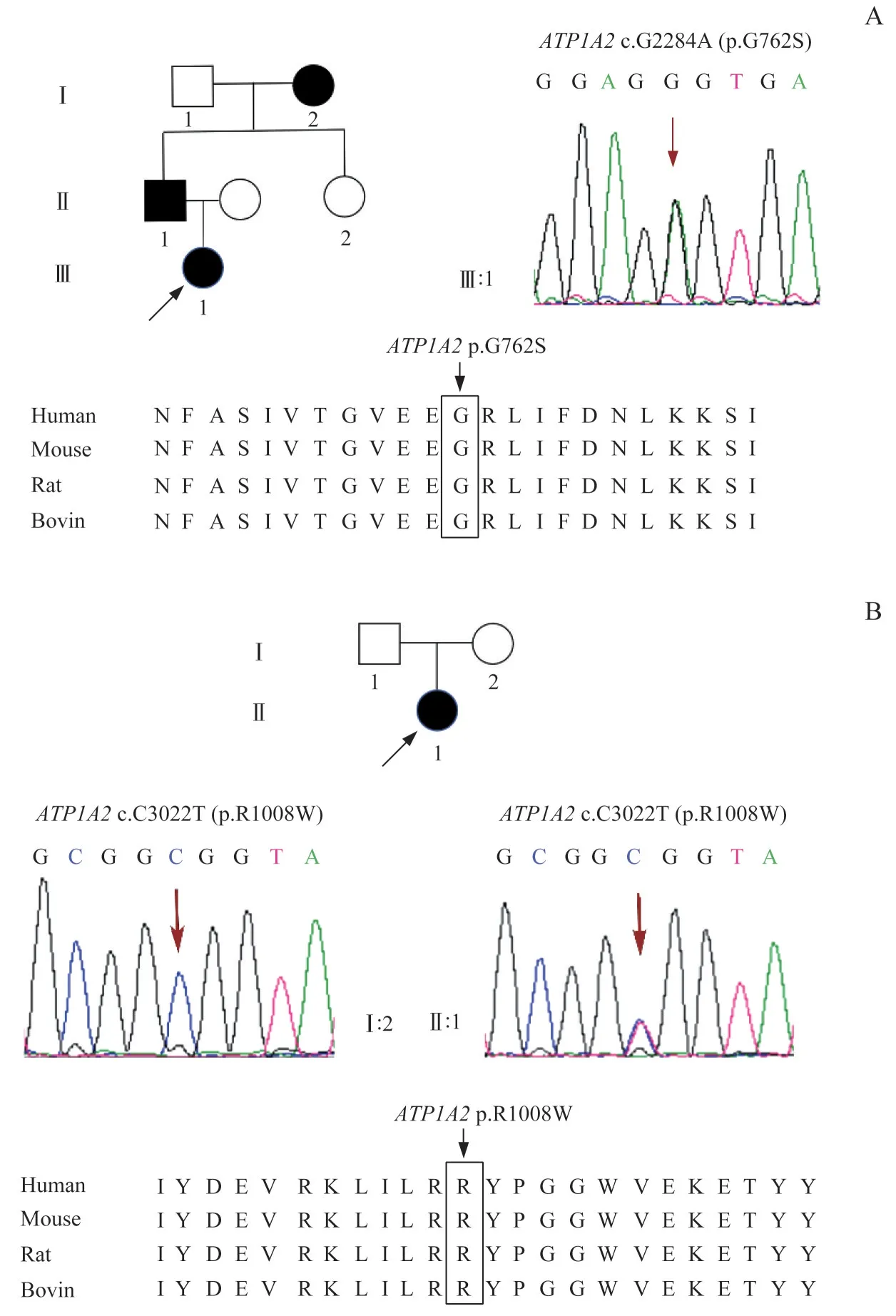
图1 ATP1A2基因突变患者家系图及ATP1A2一代测序图Fig 1 Pedigrees of ATP1A2 gene mutation patients and ATP1A2 Sanger sequencing diagram
2 讨论
本文报道的2 例患者均有偏侧肢体无力、视觉障碍等典型偏瘫型偏头痛先兆症状,后出现发作性头痛伴随恶心、呕吐,但未伴有其他神经系统疾病。根据《国际头痛疾病分类(第三版)》诊断标准,2 例患者均符合典型单纯偏瘫型偏头痛的诊断。病例1 存在ATP1A2基因的杂合突变c.G2284A/p.G762S,此前报道为致病性突变。病例2 存在ATP1A2基因的杂合变异c.C3022T/p.R1008W,既往未有该变异单独致病的个案报道,根据ACMG 指南评为“意义不明确”;后续可对其父亲进行基因共分离检测以及必要的功能实验,进一步评定其致病性。
家族性偏瘫型偏头痛呈常染色体显性遗传并且伴有不完全外显,外显率为70%~90%[8]。家族性和散发性偏瘫型偏头痛在临床表现上并无差异[3]。偏瘫型偏头痛可由外伤、感染、疲劳、情绪应激等诱发[4,8]。在先兆期可出现短暂性肢体无力以及其他先兆症状中的1 种及以上,如视觉闪烁或视觉暗点、感觉异常和/或失语[1]。肢体无力症状通常从一侧肢体的远端向近端发展,也可出现左右肢体交替,极少双侧同时累及[9-10]。各种先兆症状可能出现重叠,通常在20~60 min 后得到缓解[3]。大多数患者(98%)发作时都会出现头痛,头痛位置不固定,可为单侧或双侧,可为无力肢体的同侧或对侧[4]。但在疾病早期可能只出现轻微头痛,甚至没有头痛症状[11]。先兆症状和偏头痛出现的顺序也不固定,有二者同时出现的报道[12]。此外,该疾病临床表型的严重程度不一,可表现为仅出现先兆症状和偏头痛的单纯型[6,13],也可出现较为严重的表型,即伴随出现发热、意识障碍(严重时会出现昏迷)、认知功能障碍[14]、持续性共济失调、癫痫发作、智力低下等其他神经系统疾病以及影像学改变[15-17]。
ATP1A2基因定位于1q23上0.9 Mb区域,包含23个外显子,跨度约为25 kb[18]。其突变不仅可以导致偏瘫型偏头痛2 型[19],也可引起普通偏头痛[20]、儿童交替性偏瘫1型[21]、家族性基底型偏头痛[22]的发生。迄今,在家族性和散发性偏瘫型偏头痛病例中发现了超过90 种突变,绝大多数为错义突变[8],尚未发现基因型和表型之间的明确相关性[13]。总结国内报道的5例ATP1A2相关偏瘫型偏头痛[6,11,14,16,23],临床表现分别为单纯偏瘫型偏头痛(p.G762S)以及偏瘫型偏头痛伴急性脑病(p.E825K、 p.T378I、p.G715R、p.P782R)。癫痫发作是偏瘫型偏头痛患者最严重的症状之一[24]。在国内报道的5 例病例中,4 例合并脑病表现的患者均有癫痫发作。在该疾病发作及发作间期,部分患者的脑电图在偏瘫对侧的大脑半球可记录到异常慢波[10];当合并癫痫发作时,脑电图可能会出现癫痫样放电现象[23,25]。本文2例患者均未出现异常脑电改变。病例1 与此前p.G762S 突变导致家族性偏瘫型偏头痛的10 例患者临床表现类似[6],感染、疲劳是这2个家系共同的诱发因素。这些患者头痛发作前均出现偏瘫、视觉障碍等先兆症状,持续时间在1 h 左右。头痛主要特征为发作性单侧疼痛,且不伴有其他神经系统合并症状,提示p.G762S突变可能更易表现为单纯型。不同的是,在既往报道的10个p.G762S导致的偏瘫型偏头痛家系中,多数患者(7/10)同时出现眩晕、耳鸣、听觉减退、醉酒步态等脑干症状;而本文中病例1 并无类似症状,提示家族性偏瘫型偏头痛存在一定的临床异质性。并且,脑干症状在散发性偏瘫型偏头痛中也经常出现(76/105)[9]。因此,对于发作性眩晕、肢体乏力合并偏头痛的患者,临床上需要考虑到偏瘫型偏头痛的可能。
ATP1A2基因编码星形胶质细胞上Na+/K+泵的α2亚基,主要在中枢神经系统中表达[4]。ATP1A2基因突变通过改变Na+/K+泵的功能,抑制了神经胶质细胞对突触间隙K+和谷氨酸的再摄取,大脑产生皮质扩散抑制,表现出先兆症状[19,26-27]。α2 亚基由10 个跨膜蛋白(TM)组成,其中TM4~TM5 包含了ATP 的催化位点和离子转运通道,是Na+/K+泵的功能区域,同时也是ATP1A2基因的热点突变区域[28]。在此前单纯偏瘫型偏头痛家系的报道中,已证实病例1 患者c.G2284A 突变位于TM4~TM5 区域。c.G2284A/p.G762S 突变使蛋白正常折叠受阻,从而影响Na+/K+泵与阳离子的结合,降低催化反应速率[6]。c.C3022T/p.R1008W 突变位于TM10,即Na+/K+泵α亚基的C 端[29]。C 端主要影响泵与阳离子结合的亲和力,其中精氨酸作为电压感应器对电压依赖性的离子通道起到重要调节作用[30],但该突变对离子通道的作用机制目前尚未阐明[29]。
目前,偏瘫型偏头痛的治疗多依据小型研究和病例报道[10,27]。临床上常给予作用于钠或钙离子通道的药物进行长期预防治疗[31],以减少疾病的发作频率、严重程度和持续时间。这些药物包括维拉帕米、氟桂利嗪、乙酰唑胺,以及托吡酯、丙戊酸钠、拉莫三嗪等抗癫痫药物[3,7,10]。另外,针对偏瘫型偏头痛伴有急性重症脑病的患者,早期抑制谷氨酸的兴奋性、保护线粒体功能以及给予传统治疗偏头痛的药物,症状可以得到改善[16]。本文病例1 患者因曾有抑郁症病史,存在氟桂利嗪应用的禁忌证,故尝试采用托吡酯治疗,随访1 个月时患者无头痛以及先兆症状出现,提示治疗效果良好。
综上,偏瘫型偏头痛患者的临床症状多样,可表现为单纯型或伴有精神发育迟滞、意识障碍、癫痫等其他神经系统疾病,其临床表现存在一定异质性,临床诊断相对复杂,基因检测是明确诊断重要方法之一。本文病例1所携带的c.G2284A/p.G762S突变位于ATP1A2基因突变的热点区域,并可能更易表现为单纯偏瘫型偏头痛。病例2 患者ATP1A2基因存在的杂合突变c.C3022T/p.R1008W 国内少有报道,该突变对Na+/K+泵的功能影响有待进一步的细胞功能学研究。
利益冲突声明/Conflict of Interests
所有作者声明不存在利益冲突。
All authors disclose no relevant conflict of interests.
伦理批准和知情同意/Ethics Approval and Patient Consent
本病例报告已通过上海交通大学医学院附属第六人民医院伦理委员会的审核批准(文件号2021-219)。患者或其亲属已经签署知情同意书。
This report were reviewed and approved by the Ethics Committee of Shanghai Sixth People's Hospital,Shanghai Jiao Tong University(Approval Letter No. 2021-219,dated 09/30/2021). Consent letters have been signed by the patients or their relatives.
作者贡献/Authors'Contributions
曹立、田沃土、詹飞霞参与了资料收集;丁思奇、黄啸君参与了论文的写作和修改。所有作者均阅读并同意了最终稿件的提交。
The clinical data were collected by CAO Li,TIAN Wotu and ZHAN Feixia. The manuscript was drafted and revised by DING Siqi and HUANG Xiaojun. All the authors have read the last version of paper and consented for submission.
·Received:2022-04-29
·Accepted:2022-06-06
·Published online:2022-08-08
参·考·文·献
[1] Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders,3rd edition[J]. Cephalalgia,2018,38(1):1-211.
[2] LYKKE THOMSEN L, KIRCHMANN ERIKSEN M, FAERCH ROMER S,et al. An epidemiological survey of hemiplegic migraine[J].Cephalalgia,2002,22(5):361-375.
[3] PELZER N, STAM A H, HAAN J, et al. Familial and sporadic hemiplegic migraine: diagnosis and treatment[J]. Curr Treat Options Neurol,2013,15(1):13-27.
[4] RUSSELL M B, DUCROS A. Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics,diagnosis,and management[J]. Lancet Neurol,2011,10(5):457-470.
[5] FERRARI M D, KLEVER R R, TERWINDT G M, et al. Migraine pathophysiology:lessons from mouse models and human genetics[J].Lancet Neurol,2015,14(1):65-80.
[6] TANG W J, ZHANG M C, QIU E C, et al. A Chinese family with familial hemiplegic migraine type 2 due to a novel missense mutation inATP1A2[J]. Cephalalgia,2019,39(11):1382-1395.
[7] RICHARDS S,AZIZ N,BALE S,et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med,2015,17(5):405-424.
[8] SUTHERLAND H G,ALBURY C L,GRIFFITHS L R. Advances in genetics of migraine[J]. J Headache Pain,2019,20(1):72.
[9] THOMSEN L L,OSTERGAARD E,OLESEN J,et al. Evidence for a separate type of migraine with aura:sporadic hemiplegic migraine[J].Neurology,2003,60(4):595-601.
[10] DI STEFANO V, RISPOLI M G, PELLEGRINO N, et al.Diagnostic and therapeutic aspects of hemiplegic migraine[J].J Neurol Neurosurg Psychiatry,2020,91(7):764-771.
[11] CHEN H, SUN X L, WANG R Y, et al. A case report of atypical hemiplegic migraine with nonheadache onset in a Chinese child[J].BMC Neurol,2021,21(1):267.
[12] JEN J C, KLEIN A, BOLTSHAUSER E, et al. Prolonged hemiplegic episodes in children due to mutations inATP1A2[J].J Neurol Neurosurg Psychiatry,2007,78(5):523-526.
[13] LI Y J, TANG W J, KANG L, et al. Functional correlation ofATP1A2mutations with phenotypic spectrum: from pure hemiplegic migraine to its variant forms[J]. J Headache Pain,2021,22(1):92.
[14] 胡笑月,汤继宏,奚晓隽,等. 以发作性意识障碍为主要临床表型的偏瘫型偏头痛(附1 例报告及文献复习)[J]. 中国临床神经科学,2020,28(6):649-654.HU X Y, TANG J H, XI X J, et al. Hemiplegic migraine presenting with recurrent episodes of impaired consciousness: a case report and literature review[J]. Chin J Clin Neurosci,2020,28(6):649-654.
[15] VAN DEN MAAGDENBERG A M, TERWINDT G M, HAAN J, et al. Genetics of headaches[J]. Handb Clin Neurol,2010,97:85-97.
[16] DU Y, LI C, DUAN F J, et al. Early treatment in acute severe encephalopathy caused byATP1A2mutation of familial hemiplegic migraine type 2: case report and literature review[J].Neuropediatrics,2020,51(3):215-220.
[17] WILBUR C, BUERKI S E, GUELLA I, et al. An infant with epilepsy and recurrent hemiplegia due to compound heterozygous variants inATP1A2[J]. Pediatr Neurol,2017,75:87-90.
[18] SHULL M M, PUGH D G, LINGREL J B. Characterization of the human Na, K-ATPase α2 gene and identification of intragenic restriction fragment length polymorphisms[J]. J Biol Chem, 1989,264(29):17532-17543.
[19] DE FUSCO M, MARCONI R, SILVESTRI L, et al.Haploinsufficiency ofATP1A2encoding the Na+/K+pump α2 subunit associated with familial hemiplegic migraine type 2[J]. Nat Genet,2003,33(2):192-196.
[20] TODT U, DICHGANS M, JURKAT-ROTT K, et al. Rare missense variants inATP1A2in families with clustering of common forms of migraine[J]. Hum Mutat,2005,26(4):315-321.
[21] BASSI M T, BRESOLIN N,TONELLI A, et al. A novel mutation in theATP1A2gene causes alternating hemiplegia of childhood[J].J Med Genet,2004,41(8):621-628.
[22] AMBROSINI A,D'ONOFRIO M,GRIECO G S,et al. Familial basilar migraine associated with a new mutation in theATP1A2gene[J].Neurology,2005,65(11):1826-1828.
[23] WANG P,YANG Y R, ZHANG H B, et al. Cognitive dysfunction in a patient with migraine andAPT1A2mutation: a case report[J].Neurol Sci,2021,42(12):5425-5431.
[24] COSTA C, PRONTERA P, SARCHIELLI P, et al. A novelATP1A2gene mutation in familial hemiplegic migraine and epilepsy[J].Cephalalgia,2014,34(1):68-72.
[25] BEAUVAIS K, CAVÉ -RIANT F, DE BARACE C, et al. NewCACNA1Agene mutation in a case of familial hemiplegic migraine with status epilepticus[J]. Eur Neurol,2004,52(1):58-61.
[26] CAPUANI C,MELONE M,TOTTENE A,et al. Defective glutamate and K+clearance by cortical astrocytes in familial hemiplegic migraine type 2[J]. EMBO Mol Med,2016,8(8):967-986.
[27] MARTÍNEZ E, MORENO R, LÓPEZ-MESONERO L, et al.Familial hemiplegic migraine with severe attacks: a new report withATP1A2mutation[J]. Case Rep Neurol Med,2016,2016:3464285.
[28] FRIEDRICH T, TAVRAZ N N, JUNGHANS C.ATP1A2mutations in migraine: seeing through the facets of an ion pump onto the neurobiology of disease[J]. Front Physiol,2016,7:239.
[29] SWEADNER K J, ARYSTARKHOVA E, PENNISTON J T, et al.Genotype-structure-phenotype relationships diverge in paralogsATP1A1,ATP1A2,andATP1A3[J]. Neurol Genet,2019,5(1):e303.
[30] MORTH J P, PEDERSEN B P, TOUSTRUP-JENSEN M S, et al.Crystal structure of the sodium-potassium pump[J]. Nature, 2007,450(7172):1043-1049.
[31] VGONTZAS A, BURCH R. Episodic migraine with and without aura: key differences and implications for pathophysiology,management, and assessing risks[J]. Curr Pain Headache Rep, 2018,22(12):78.
