肿瘤微环境中免疫细胞的代谢研究进展
2022-10-21林家俞秦洁洁蒋玲曦
林家俞,秦洁洁,蒋玲曦
上海交通大学医学院附属瑞金医院普外科胰腺疾病诊疗中心,上海交通大学医学院胰腺疾病研究所,上海 200025
肿瘤微环境是一个复杂的、具有高度异质性的动态综合系统,主要由肿瘤细胞、肿瘤相关成纤维细胞(cancer-associated fibroblasts,CAFs)、肿瘤相关免疫细胞和微血管等成分组成。
多年的研究证明,代谢途径对肿瘤微环境中肿瘤细胞的增殖、转移与免疫逃逸具有重要作用[1]。在肿瘤发生与进展过程中,糖酵解的激活、脂质代谢的增加、线粒体生物合成增强等代谢途径重塑了局部肿瘤微环境,改变了肿瘤微环境中免疫细胞的代谢适应性[2]。肿瘤微环境中细胞固有代谢紊乱共同导致营养物质的消耗、环境pH酸化、缺氧及调节性代谢产物的产生,进而影响抗肿瘤免疫反应,强化肿瘤细胞对免疫治疗的抵抗,促进免疫检查点分子的过度表达和肿瘤转移[3-4]。
肿瘤微环境中免疫细胞的代谢状态是影响其发挥正常免疫应答的关键因素,因此本综述拟总结肿瘤微环境中主要免疫细胞的代谢途径,概括参与肿瘤免疫反应的免疫细胞的代谢特征,归纳免疫细胞代谢途径改变的分子机制,以期为通过靶向免疫细胞代谢途径提高肿瘤免疫疗效提供新思路。
1 肿瘤免疫微环境
浸润到肿瘤内部的淋巴细胞介导了免疫抑制的肿瘤微环境,帮助肿瘤细胞实现免疫逃逸,促进肿瘤的恶性发展。肿瘤免疫微环境由一系列不同的细胞类型构成(图1),包括T 淋巴细胞、B 淋巴细胞、肿瘤相关巨噬细胞(tumor-associated macrophages,TAMs)、自然杀伤细胞(natural killer cells,NKs)、树突状细胞(dendritic cells,DCs)、肿瘤相关中性粒细胞(tumor-associated neutrophils,TANs)和髓源性抑制细胞(myeloid-derived suppressor cells,MDSCs)等。同时,免疫细胞代谢途径的改变使其功能具有两面性:一方面,浸润至肿瘤内部的免疫细胞在肿瘤入侵的初始阶段发挥抗肿瘤作用;另一方面,在肿瘤进展过程中,部分免疫细胞逐渐转变成促肿瘤表型,发挥免疫抑制功能,协助肿瘤免疫逃逸及远处转移[5]。
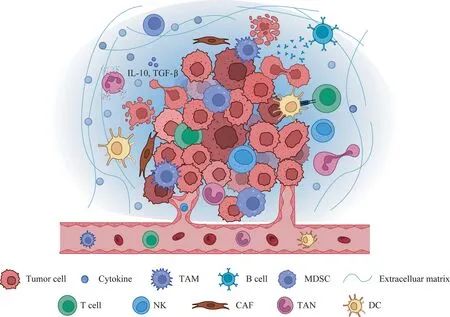
图1 肿瘤免疫微环境主要的免疫细胞Fig 1 Main immune cells in tumor immune microenvironment
由癌细胞代谢异常释放的各种生化分子重塑肿瘤微环境,影响免疫细胞的正常免疫反应。代谢串扰是癌细胞在不利条件下维持生长的一种方式。癌细胞可通过释放犬尿氨酸和乳酸等机制逃避细胞毒性T淋巴细胞介导的杀伤作用,从而增强调节性T细胞(regulatory T cells,Tregs)和MDSCs的免疫抑制功能[6]。此外,癌细胞通过对营养物质的恶性竞争实现快速增长的目的,如:通过Warburg效应满足快速增殖的生物能量和生物合成需求[7]。因此,肿瘤细胞的代谢多样性将肿瘤微环境塑造为一个酸性、缺氧、营养物质匮乏的场所,进而影响免疫细胞的代谢途径,限制免疫细胞的抗肿瘤效应[8]。
2 固有免疫细胞代谢
2.1 TAMs
巨噬细胞是重要的固有免疫细胞,主要通过吞噬及消化细胞碎片和病原体,并激活其他免疫细胞对抗病原体入侵来发挥其功能。而浸润在肿瘤组织中的TAMs 具有高度的可塑性和异质性[9]。肿瘤早期,Toll 样受体(toll-like receptors,TLR)激动剂等促炎细胞因子可促进TAM 向M1 型极化,M1 型巨噬细胞产生的一氧化氮(nitric oxide,NO) 和活性氧(reactive oxygen species,ROS)可显著抑制肿瘤细胞增殖,杀死肿瘤细胞[10]。肿瘤进展期,白细胞介素-4(interleukin-4,IL-4)与集落刺激因子1(colony stimulating factor 1,CSF1)诱导TAMs向M2型极化,M2 型巨噬细胞分泌表皮细胞生长因子(epidermal growth factor,EGF)、基质金属蛋白 酶-9 (matrix metalloprotein 9,MMP-9)等蛋白抑制抗肿瘤效应,促进肿瘤的进展[11]。
2 种极化形式的巨噬细胞表现出不同的代谢模式。M1 型巨噬细胞激活后,主要依赖于糖酵解代谢途径供能,并通过磷酸戊糖途径(glycolysis pentose phosphate pathway,PPP)产生ROS,发挥抗肿瘤效应[12-13]。肿瘤微环境缺氧环境可诱导M1 型巨噬细胞的 缺 氧 诱 导 因 子1α (hypoxia inducible-factor 1α,HIF1α)激活上调,诱导糖酵解基因如葡萄糖转运体1 (glucose transporter 1,GLUT1)、 己 糖 激 酶2(hexokinase 2,HK2)过度表达。同时,雷帕霉素靶蛋白信号(mechanistic target of rapamycin,mTOR)通过NOR 样受体热蛋白结构域相关蛋白3(NODlike receptor thermal protein domain associated protein 3,NLRP3)炎症小体以己糖激酶依赖性方式促使M1 型细胞完成糖酵解获取能量[14]。此外,M1 型巨噬细胞还可通过核因子κB(nuclear factor κB,NFκB)途径下调碳水化合物激酶样蛋白(carbohydrate kinase-like protein,CARKL)增加PPP 的代谢水平,产生大量还原型辅酶Ⅱ(nicotinamide adenine dinucleotide phosphate,NADPH)。NADPH 不仅可用于维持其自身氧化还原的平衡以及生物合成代谢,还可进一步促进ROS和NO的产生[15]。
相反,肿瘤微环境中发挥免疫抑制功能的M2 型巨噬细胞依赖于氧化磷酸化 (oxidative phosphorylation,OXPHOS)代谢途径供能[16]。而脂肪酸氧化(fatty acid oxidation,FAO)也是其供能途径,该过程以低水平糖酵解为代价,高表达CD36,上调FAO 水平,从而促进线粒体OXPHOS 过程[17]。此外,M2 型细胞合成大量精氨酸酶(arginase,ARG)和吲哚胺2,3-双加氧酶1(indoleamine 2,3-dioxygenase 1,IDO1),消耗精氨酸与色氨酸,导致免疫功能障碍[18]。肿瘤微环境中的细胞因子也可以影响TAMs的分化与功能。CSF1是TAMs发挥免疫抑制功能的一个重要细胞因子。肿瘤细胞在快速增殖过程中释放大量的CSF1 至细胞外,CSF1 可与TAMs 上的受体结合,招募其至肿瘤细胞周围,诱导其FAO水平上升,促使其极化为M2型巨噬细胞[19]。
2.2 TANs
中性粒细胞来源于髓系祖细胞,在骨髓中发育成熟后,在细胞因子IL-8的趋化下,其释放至血液参与固有免疫,调节适应性免疫。肿瘤微环境中,一方面在干扰素-β(interferon-β,IFN-β)、肿瘤坏死因子-α(tumor necrosis factor,TNF-α)作用下,TANs 向具有高免疫活性的抗肿瘤N1 型极化,促进CD8+T 细胞活化;另一方面,转化生长因子-β(transforming growth factor-β,TGF-β)、IL-8 等促使TANs 向促肿瘤的N2 型极化。N2 型细胞释放ARG1、组织蛋白酶、促血管生成细胞因子等,抑制肿瘤免疫,加快肿瘤进展。
TANs 依赖于糖酵解代谢途径供能[20]。TANs 中GLUT1表达水平上调,但在敲除GLUT1基因后,TANs 摄取葡萄糖的能力下降,存活率显著降低[21]。PPP 所生成的NADPH 是NADPH 氧化酶的辅助因子,可以辅助TANs发挥抗肿瘤免疫功能。研究[22]表明,肿瘤细胞通过干细胞因子(stem cell factor,SCF)/c-Kit信号转导诱导TANs产生氧化表型,增强其线粒体的OXPHOS功能,使得TANs即使在葡萄糖不足的条件下,也可通过FAO 来维持NADPH 的产生,维持ROS水平,发挥免疫抑制功能。
2.3 NKs
NKs作为人体抵抗肿瘤的第一道防线,可在靶细胞表面释放穿孔素导致细胞穿孔,使颗粒酶b 进入肿瘤细胞诱导凋亡,从而非特异性杀伤肿瘤细胞。这个过程既不需要抗原致敏与抗体参与,也无组织相容性复合体(major histocompatibility complex,MHC)限制,并且还能通过分泌细胞因子,促进适应性免疫细胞的抗肿瘤作用。
NKs 活化后,胞内的固醇调节元件结合蛋白(sterol regulatory element- binding protein,SREBP)与雷帕霉素靶蛋白复合物1 (mechanistic target of rapamycin complex 1,mTORC1)表达上调,增强有氧糖酵解与OXPHOS 代谢途径水平[23]。转录因子c-Myc 可显著提高NKs 的代谢水平;若c-Myc 蛋白缺陷,NKs将减少糖代谢关键酶和线粒体酶的表达,导致免疫功能发生障碍[24]。乳酸能干扰NKs 的代谢。乳酸在肿瘤微环境的积累使NKs 胞内pH 降低,导致pH 依赖性线粒体的应激反应与代谢功能障碍,ROS不断累积,促使NKs 发生凋亡[25]。过度脂质代谢也会影响NKs 的正常功能。脂质转运蛋白表达上调导致细胞过度摄取脂肪酸,从而激活过氧化物酶体增殖激活受体(peroxisome proliferators-activated receptor,PPAR)-γ/PPAR-δ信号,抑制NKs的代谢活性并产生细胞毒性[26]。
2.4 DCs
DCs在固有免疫识别病原体、启动适应性免疫细胞活化中发挥关键作用。在肿瘤微环境中,DCs接收并整合由细胞因子、损伤相关分子模式(damageassociated molecular patterns,DAMPs) 等受体感知的环境信号,通过MHC 分子提呈抗原肽,进一步诱导T细胞活化和分化,启动适应性免疫反应,同时还可分泌细胞因子和生长因子,增强T 细胞、NKs等免疫细胞活性,建立完整的抗肿瘤免疫反应。
不同时期的DCs 代谢状态不一。未成熟的DCs主要依赖于OXPHOS 代谢途径;当TLR 激活触发DCs活化及发育成熟后,DCs的代谢途径从OXPHOS转变为糖酵解,以支持其代谢需求与抗原提呈功能[27]。 磷 酸 肌 醇 3- 激 酶- 蛋 白 激 酶 B(phosphatidylinositol 3 kinase-protein kinase B,PI3KAKT)、TANK 结 合 激 酶1 (tank binding kinase 1,TBK1)和κB 激酶ε 抑制剂(inhibitor of κB kinase ε,IKKε)通路信号驱动DCs 糖酵解代谢。随着DCs 活化,一氧化氮合酶2(nitric oxide synthase 2,NOS2)诱导生成的NO降低OXPHOS代谢水平,阻止细胞色素C氧化酶活化,并激活PI3K-AKT途径,抑制AMP依赖的蛋白激酶途径(adenosine 5'-monophosphate activated protein kinase,AMPK)[28]。
细胞因子可调节DCs的生物学功能。肿瘤细胞通过大量释放巨噬细胞清道夫受体1 (macrophage scavenger receptor 1,MSR1)等细胞因子,使得DCs胞内脂质异常积累,发生抗原提呈功能障碍,降低对T细胞的诱导活化功能[29]。而通过靶向MSR1、乙酰辅酶A 羧化酶(acetyl CoA carboxylase,ACC)或Xbox 结合蛋白1(X-box binding protein 1,XBP1)可调节DCs胞内的脂质积累,恢复DCs的免疫活性[30]。
2.5 MDSCs
MDSCs 是由未成熟的,无法分化为粒细胞、巨噬细胞和DCs的髓系祖细胞组成的免疫抑制性先天细胞群。生长因子如粒细胞集落刺激因子(granulocyte colony stimulating factor,G-CSF)和血管内皮生长因子(vascular endothelial growth factor,VEGF)可促进MDSCs 增殖,并抑制其最终分化成巨噬细胞、DCs或粒细胞;而促炎细胞因子如IL-4和IL-6则可促进MDSCs 的病理活化。MDSCs 通过释放NO 与ROS,产生IL10、IFN-γ 等免疫调节细胞因子等方式抑制T细胞活性。
在缺氧、营养物质匮乏的肿瘤微环境中,MDSCs 通过糖代谢与OXPHOS 代谢途径适应环境,维持免疫抑制作用[31]。研究[32]表明,肿瘤微环境中的糖酵解中间代谢物磷酸烯醇丙酮酸(phosphoenolpyruvate,PEP)可防止过量ROS 产生,保护MDSCs逃脱ROS诱导的细胞凋亡,促进MDSCs存活与分化。脂质代谢可调节MDSCs 的免疫功能。肿瘤微环境中的乳酸增强了MDSCs 的由CD36 介导的脂质摄取能力,使FAO 替代糖酵解成为MDSCs 的主要能量来源。CD36缺失或FAO抑制会破坏MDSCs免疫抑制功能,提高化学治疗和免疫治疗的效果,延缓肿瘤生长[33]。
3 适应性免疫细胞代谢
3.1 T细胞
CD4+或CD8+T 细胞通过表达T 细胞受体(T cell receptor,TCR)α/β,识别肿瘤抗原和自身抗原,在癌症或自身免疫性疾病的免疫反应过程中发挥关键作用。经同源抗原刺激后,初始CD8+T 细胞分化为细胞毒性效应细胞和长期记忆细胞,初始CD4+T 细胞分化为辅助性T 细胞1(hepler T cell 1,Th1)、Th2、Th17、滤泡辅助性T 细胞(follicular helper T cell,Tfh)、Tregs 和长期记忆细胞。其中,Tregs 是一类以高表达叉头框蛋白P3 (forkhead box protein P3,FOXP3)、CD25、CD4 为主要特征的免疫抑制性T 细胞。Tregs 常在肿瘤中积聚,通过脂质代谢与OXPHOS 途径为免疫反应供能,维持肿瘤微环境的免疫抑制环境,促进肿瘤浸润与转移[34-35]。
在肿瘤免疫微环境中,一方面免疫受体、信号蛋白和转录因子等会促进T细胞的活化,另一方面代谢途径改变会影响T细胞存活、增殖、分化、功能发挥等重要生物学过程[36]。基于T 细胞作为抗肿瘤免疫中的关键免疫细胞,下面从葡萄糖代谢、脂质代谢、氨基酸代谢、线粒体调控4 个代谢途径来阐述肿瘤微环境中T细胞的代谢特征。
3.1.1 葡萄糖代谢 TCR 识别肿瘤抗原后,在CD28介导的共刺激下,激活PI3K-AKT-mTOR 通路,上调HIF1α 和c-Myc 等转录因子的表达,增加GLUT1 蛋白、代谢酶和氨基酸转运蛋白的表达,增强糖酵解与谷氨酰胺代谢水平[37-38]。在肿瘤微环境中,葡萄糖的匮乏抑制TCR 依赖性Ca2+和NFAT 信号的激活,使得CD8+T 细胞无法正常分化,从而导致抗肿瘤功能障碍[39]。而且,糖酵解的关键酶之一烯醇化酶-1(enolase-1,ENO-1)可调节FOXP3外显子2 剪接变体的表达,诱导Tregs 分化与功能的发挥[40]。此外,乳酸是一个调节T细胞增殖、功能发挥等生物学行为的重要因素。乳酸通过阻止C-X-C 趋化因子受体3(C-X-C motif chemokine receptor 3,CXCR3)与其配体结合,抑制Th 细胞的功能,降低抗肿瘤免疫活性[41]。单羧酸转运体(monocarboxylate transporter,MCT)是乳酸的转运体,Tregs 可通过MCT1 摄取肿瘤微环境中的乳酸,将乳酸转化为苹果酸与柠檬酸后转移至线粒体中参与三羧酸循环(tricarboxylic acid cycle,TCA),增强程序性死亡受体1(programmed cell death protein 1,PD-1)表达与Treg的免疫抑制功能,降低免疫治疗效果[42-43]。
3.1.2 脂质代谢 脂质是一种不可或缺的营养物质。调节细胞脂质合成与胆固醇摄取的关键蛋白固醇调节元件结合蛋白1 (sterol regulatory element-binding protein,SREBP1)和SREBP2 在T 细胞的细胞膜合成过程中发挥重要作用。缺失SREBP1 与SREBP2 显著抑制CD8+T 细胞的增殖、代谢与抗肿瘤活性[44]。此外, 乙酰辅酶A 乙酰转移酶1 (acetyl-CoA acetyltransferase 1,ACAT1)是一种关键的胆固醇酯化酶,抑制ACAT1 的表达可降低CD8+T 细胞胆固醇酯化,增强TCR 的聚集和信号转导,提高T 细胞的增殖与抗肿瘤活性[45]。脂肪酸代谢可影响T 细胞的发育、分化和功能的发挥。CD36 是Tregs 中维持线粒体呼吸的一个关键因素。Tregs 高表达CD36,增强长链脂肪酸的摄取能力,维持PPAR-β 信号依赖性线粒体的适应性[46]。mTORC1 信号通路可增强Tregs 中的脂质和胆固醇代谢,提高Tregs 对细胞外游离脂肪酸的摄取, 上调诱导共刺激分子(inducible co-stimulator,ICOS)等细胞增殖和免疫抑制相关基因的表达,发挥免疫抑制功能[47]。肿瘤微环境中, 肿瘤细胞高表达的前列腺素E2(prostaglandin E2,PGE2)可增加Tregs 特异性转录因子FOXP3 的表达,促进初始T 细胞分化成Tregs,增强肿瘤免疫抑制作用[48]。
3.1.3 氨基酸代谢 氨基酸是细胞维持高代谢水平所必需的营养物质之一。T 细胞依赖氨基酸代谢途径合成自身所需蛋白质与核苷酸。色氨酸是一种必需氨基酸,其在肿瘤微环境的浓度决定了T细胞的反应强度和抗肿瘤效应的有效性。肿瘤细胞高表达的IDO可降解色氨酸为犬尿氨酸,导致肿瘤微环境中色氨酸缺乏。一般性调控阻遏蛋白激酶2 (general control nonderepressible 2,GCN2)是一种应激反应激酶,参与调控多种生物学过程。色氨酸的缺失可激活CD8+T 细胞中的GCN2,下调CD3ζ 链,导致CD8+T细胞周期停滞和细胞毒效应受损[49]。L-精氨酸是T细胞生命周期中所必需的一种氨基酸,不仅能诱导T细胞由糖酵解向OXPHOS 转变,还能进一步促进T细胞存活与记忆T细胞产生[50]。甲硫氨酸也是调节T细胞功能的一种重要氨基酸。肿瘤细胞通过上调甲硫氨酸转运蛋白的表达水平,增加甲硫氨酸的摄取能力,降低肿瘤微环境中的甲硫氨酸水平,减少甲基供体,从而影响CD8+T细胞组蛋白H3亚基第79位赖氨酸(H3K79)的甲基化程度,造成CD8+T 细胞的抗肿瘤功能障碍[51]。
3.1.4 线粒体调控 线粒体对T 细胞的调控至关重要。TCR 可增强线粒体的生物活性和代谢重构,这个过程是满足T 细胞活化和代谢要求所必需的[52]。肿瘤微环境的缺氧环境可通过下调MYC表达水平来促进线粒体结构损伤并减少ATP 的产生,从而诱导T细胞耗竭(T cell exhaustion,TExh),使CD8+T 细胞发生抗肿瘤功能障碍[53]。另外,在CD8+T 细胞由效应性T细胞向记忆性T细胞转化过程中,SENP1-Sirt3轴的激活可显著降低CD8+T 细胞线粒体中的乙酰化水平,增加线粒体融合和OXPHOS 代谢水平,增强记忆性T 细胞的形成和与存活,促进抗肿瘤免疫活性[54]。过氧化物激活受体1α(PPAR γ coactivator-1α,PGC1α)是线粒体生物发生的关键调控因子。在免疫抑制微环境中,CD8+T 细胞内线粒体处于功能抑制状态,而增强CD8+T 细胞中PGC1α 的表达,可促进CD8+T 细胞中线粒体的生物合成,挽救线粒体功能,逆转线粒体功能抑制状态,增强抗肿瘤效应[55]。
3.2 B细胞
近些年来,肿瘤微环境中T细胞的肿瘤免疫作用已被广泛报道,但有关B 细胞的研究则较少。B 细胞在B 细胞受体(B cell receptor,BCR)的激活下活化为浆细胞,浸润于肿瘤内部,产生的大量细胞因子与抗体可通过驱动抗体依赖性细胞毒性(antibodydependent cell-mediated cytotoxicity,ADCC)和吞噬作用以及补体激活来发挥抗肿瘤免疫作用[56]。此外,B 细胞可促进肿瘤相关三级淋巴结构(tertitary lymphoid structures,TLS)的形成。TLS 支持肿瘤特异性B细胞的进一步成熟和亚型转换,促进肿瘤特异性T细胞反应的进展[57]。
与T细胞类似,经BCR刺激后,幼稚B细胞的葡萄糖摄取增加,而且位于生发中心的B细胞糖酵解活性明显增强[58]。OXPHOS对B细胞的活化至关重要。虽然已有研究[59]表明活化的B 细胞会增加葡萄糖的摄取,以满足自身需要,但在缺乏葡萄糖条件下,B细胞的生长与功能未受影响。而使用寡霉素抑制OXPHOS 代谢途径将明显抑制B 细胞的生长和分化。截至目前,对调控B细胞代谢的具体分子机制仍不清楚,还需进一步的研究。
4 基于免疫细胞代谢的药物研究
在肿瘤免疫微环境影响下,免疫细胞自发产生耐受表型相关的代谢适应,如T细胞在肿瘤微环境中主要依赖有氧糖酵解和谷氨酰胺分解代谢来供能。抑制代谢适应可提高免疫细胞的抗肿瘤效应,因此,基于肿瘤微环境的代谢特征,以糖酵解、氨基酸与脂质代谢途径为靶向的治疗可恢复抗肿瘤免疫细胞的功能,增强抗肿瘤效应。
肿瘤细胞在肿瘤微环境中通过糖酵解不断积累乳酸,从而激活免疫细胞和内皮细胞上的G蛋白偶联受体81(G-protein-coupled receptor 81,GPR81),促进血管生成和肿瘤细胞的免疫逃逸[60]。MCT1与MCT4抑制剂可增加细胞内乳酸水平,减少糖酵解,促进肿瘤细胞的死亡,并可增加T 细胞IL-2 和IFN-γ 分泌,表明MCT 抑制剂可降低肿瘤细胞增殖并促进T 细胞活化[61]。目前,MCT的抑制剂AZD3965正在进行Ⅰ期临床试验(NCT01791595),但在试验中发现,晚期肿瘤患者口服后,尿乳酸水平升高,并且出现剂量依赖性的可逆的视网膜功能障碍[62]。未来,AZD3965的临床疗效与安全性还需进一步的验证。
氨基酸是细胞代谢过程中不可或缺的部分,其中谷氨酰胺可作为靶点调节肿瘤进展和免疫反应。在乳腺癌模型中,应用谷氨酰胺抑制剂JHU-083可降低肿瘤微环境中CSF 水平,减少MDSCs 的募集,并促进其向M1 型巨噬细胞转化,减缓肿瘤生长,抑制肿瘤和MDSCs 的IDO 表达,导致犬尿氨酸水平下降[63]。在小鼠结直肠癌模型中,JHU-083 通过抑制谷氨酰胺代谢,提高CD8+T 细胞的活性,增强抗肿瘤免疫效应,诱导肿瘤消退,提高小鼠存活率[64]。目前,靶向谷氨酰胺代谢的DRP-104与靶向程序性死亡受体配体1(programmed death ligand 1,PD-L1)的阿特珠单抗联合用药现在已进入临床试验(NCT04471415),用于晚期非小细胞型肺癌与头颈部鳞状细胞癌患者的治疗。
肿瘤细胞依赖于脂肪酸从头合成来供给能量,因此,脂肪酸合酶(fatty acid synthase,FASN)可视为潜在的治疗靶点。 再刺激诱导的细胞死亡(restimulation-induced cell death,RICD)是TCR 重新激活后在效应T 细胞中触发的凋亡途径。在抑制FASN 的条件下,肿瘤微环境中的T 细胞可避免因反复受到TCR 刺激而发生RICD,从而增强T 细胞的抗肿瘤作用[65]。TVB-2640是迄今第一个进入临床研究的FASN 抑制剂,正处于多项实体瘤的临床试验中,如非小细胞型肺癌(NCT03808558)、结直肠癌(NCT02980029) 等 。 目 前 的 临 床 研 究(NCT02223247)[66]表明,TVB-2640 与紫杉醇联合疗法在多种肿瘤类型如KRAS突变的非小细胞型肺癌、卵巢癌和乳腺癌中均有疗效,部分患者可获得完全缓解或部分缓解。
对免疫细胞代谢途径的深入研究有助于更好地理解肿瘤免疫微环境内的代谢。表1 总结了肿瘤免疫微环境中主要免疫细胞的代谢途径,但是针对单一代谢酶或转运体无法从整体水平来靶向肿瘤免疫治疗。靶向免疫细胞代谢途径的治疗、放射治疗、化学治疗以及免疫治疗之间更深入的联合运用,将会取得更好的临床效果。
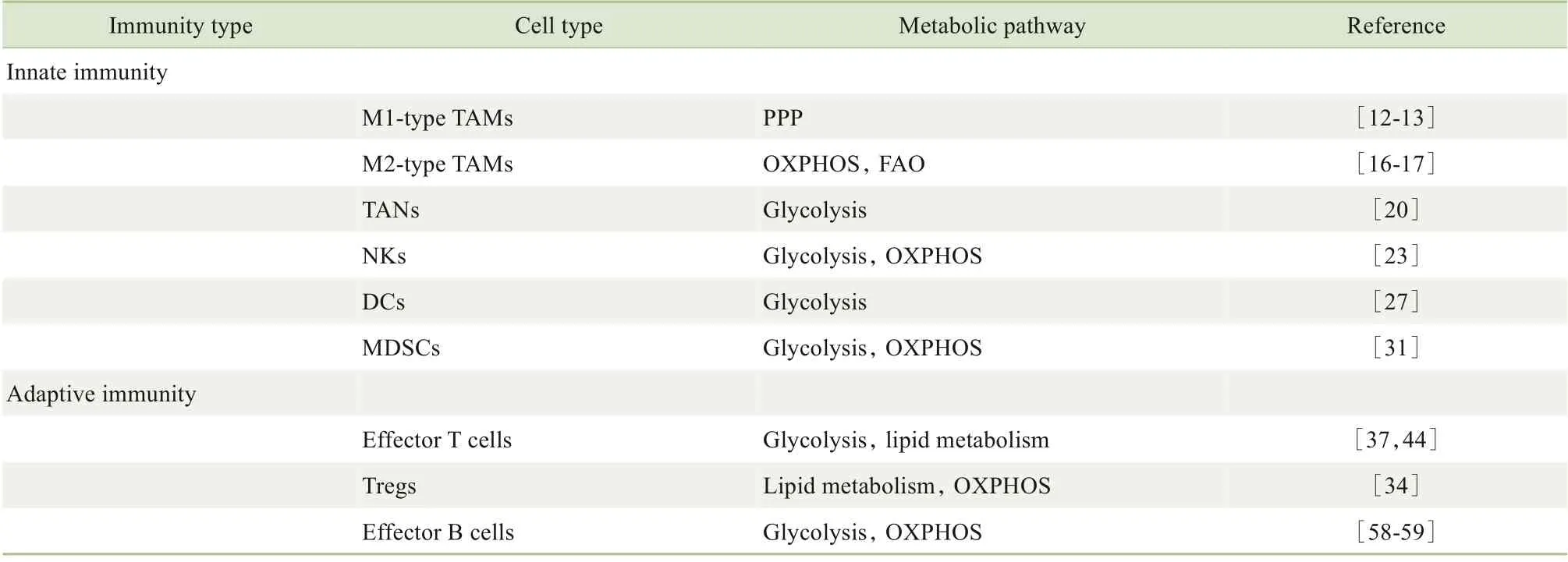
表1 肿瘤免疫微环境中主要的免疫细胞及其主要的代谢途径Tab 1 Main immune cells and their main metabolic pathways in the tumor immune microenvironment
5 结语与展望
肿瘤微环境作为肿瘤细胞赖以生存的基础,为肿瘤细胞的增殖、转移、侵袭及其他生命活动提供物质基础。肿瘤微环境的免疫状态是影响肿瘤进展的重要因素。肿瘤细胞可以通过营养竞争、分泌细胞因子及释放代谢产物等途径塑造免疫抑制的微环境,调控免疫细胞的代谢,进一步影响免疫细胞发育、分化与功能发挥,使其往促肿瘤型转化,极大限制了抗肿瘤免疫活性,这个过程有助于促进肿瘤细胞本身的免疫逃逸。因此,研究肿瘤微环境下免疫细胞通过特定代谢途径获取足够的能量以维持其抗肿瘤活性的机制就显得尤为重要。
目前,基于肿瘤微环境代谢的研究,可以聚焦免疫抑制的肿瘤微环境中免疫细胞的代谢需求,靶向效应免疫细胞代谢,选择性调节免疫细胞的极化与效应功能,将免疫应答从促肿瘤型转化为抑肿瘤型;同时与抗肿瘤及针对多靶点的免疫治疗药物联合应用,可避免适应性耐药,并显著改善肿瘤的预后和生存。未来,对于肿瘤及其内部免疫微环境层面的适应性代谢改变的进一步研究和认识可能会发现新的高特异性的靶点,而且,揭示肿瘤微环境细胞间相互作用与驱动代谢变化的分子机制,实现相关研究的临床转化,对增强肿瘤免疫治疗效果具有重要意义。
利益冲突声明/Conflict of Interests
所有作者声明不存在利益冲突。
All authors disclose no relevant conflict of interests.
作者贡献/Authors'Contributions
林家俞、秦洁洁参与了论文的写作和修改;蒋玲曦是项目的构思者及负责人,指导论文写作。所有作者均阅读并同意了最终稿件的提交。
The manuscript was drafted and revised by LIN Jiayu and QIN Jiejie;JIANG Lingxi was the conceptualizer and leader of the project and guided the writing of the paper. All the authors have read the last version of paper and consented for submission.
·Received:2022-04-27
·Accepted:2022-07-27
·Published online:2022-08-28
参·考·文·献
[1] DEBERARDINIS R J, LUM J J, HATZIVASSILIOU G, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation[J]. Cell Metab,2008,7(1):11-20.
[2] GUERRA L, BONETTI L, BRENNER D. Metabolic modulation of immunity: a new concept in cancer immunotherapy[J]. Cell Rep,2020,32(1):107848.
[3] DOMBLIDES C,LARTIGUE L,FAUSTIN B. Control of the antitumor immune response by cancer metabolism[J]. Cells,2019,8(2):104.
[4] BISWAS S K. Metabolic reprogramming of immune cells in cancer progression[J]. Immunity,2015,43(3):435-449.
[5] FAUBERT B, SOLMONSON A, DEBERARDINIS R J. Metabolic reprogramming and cancer progression[J]. Science, 2020, 368(6487):eaaw5473.
[6] LEONE R D,POWELL J D. Metabolism of immune cells in cancer[J].Nat Rev Cancer,2020,20(9):516-531.
[7] WARD P S, THOMPSON C B. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate[J]. Cancer Cell,2012,21(3):297-308.
[8] CHANG C H, QIU J, O'SULLIVAN D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression[J].Cell,2015,162(6):1229-1241.
[9] BISWAS S K, ALLAVENA P, MANTOVANI A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions[J]. Semin Immunopathol,2013,35(5):585-600.
[10] MANTOVANI A, ALLAVENA P. The interaction of anticancer therapies with tumor-associated macrophages[J]. J Exp Med, 2015,212(4):435-445.
[11] MANTOVANI A, MARCHESI F, MALESCI A, et al. Tumourassociated macrophages as treatment targets in oncology[J]. Nat Rev Clin Oncol,2017,14(7):399-416.
[12] DAI X M,LU L S,DENG S K,et al. USP7 targeting modulates antitumor immune response by reprogramming tumor-associated macrophages in lung cancer[J]. Theranostics, 2020, 10(20): 9332-9347.
[13] QING J N, ZHANG Z Z, NOVÁK P, et al. Mitochondrial metabolism in regulating macrophage polarization: an emerging regulator of metabolic inflammatory diseases[J]. Acta Biochim Biophys Sin(Shanghai),2020,52(9):917-926.
[14] MOON J S, HISATA S, PARK M A, et al. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation[J].Cell Rep,2015,12(1):102-115.
[15] HASCHEMI A, KOSMA P, GILLE L, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism[J]. Cell Metab,2012,15(6):813-826.
[16] VATS D, MUKUNDAN L, ODEGAARD J I, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation[J]. Cell Metab,2006,4(1):13-24.
[17] SU P, WANG Q, BI E G, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages[J]. Cancer Res, 2020, 80(7): 1438-1450.
[18] BANTUG G R, GALLUZZI L, KROEMER G, et al. The spectrum of T cell metabolism in health and disease[J]. Nat Rev Immunol,2018,18(1):19-34.
[19] RUFFELL B, COUSSENS L M. Macrophages and therapeutic resistance in cancer[J]. Cancer Cell,2015,27(4):462-472.
[20] RODRÍGUEZ-ESPINOSA O, ROJAS-ESPINOSA O, MORENOALTAMIRANO M M B,et al. Metabolic requirements for neutrophil extracellular traps formation[J]. Immunology,2015,145(2):213-224.
[21] ANCEY P B, CONTAT C, BOIVIN G, et al. GLUT1 expression in tumor-associated neutrophils promotes lung cancer growth and resistance to radiotherapy[J]. Cancer Res,2021,81(9):2345-2357.
[22] RICE C M, DAVIES L C, SUBLESKI J J, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression[J]. Nat Commun,2018,9(1):5099.
[23] ISAACSON B, MANDELBOIM O. Sweet killers: NK cells need glycolysis to kill tumors[J]. Cell Metab,2018,28(2):183-184.
[24] LOFTUS R M, ASSMANN N, KEDIA-MEHTA N, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice[J]. Nat Commun,2018,9(1):2341.
[25] HARMON C, ROBINSON M W, HAND F, et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liverresident NK cells in colorectal liver metastasis[J]. Cancer Immunol Res,2019,7(2):335-346.
[26] MICHELET X, DYCK L, HOGAN A, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses[J]. Nat Immunol,2018,19(12):1330-1340.
[27] KRAWCZYK C M, HOLOWKA T, SUN J, et al. Toll-like receptorinduced changes in glycolytic metabolism regulate dendritic cell activation[J]. Blood,2010,115(23):4742-4749.
[28] EVERTS B, AMIEL E, HUANG S C C, et al. TLR-driven early glycolytic reprogrammingviathe kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation[J]. Nat Immunol,2014,15(4):323-332.
[29] HERBER D L, CAO W, NEFEDOVA Y, et al. Lipid accumulation and dendritic cell dysfunction in cancer[J]. Nat Med, 2010, 16(8):880-886.
[30] CUBILLOS-RUIZ J R, SILBERMAN P C, RUTKOWSKI M R, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis[J]. Cell, 2015, 161(7): 1527-1538.
[31] HOSSAIN F, AL-KHAMI A A, WYCZECHOWSKA D, et al.Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies[J]. Cancer Immunol Res,2015,3(11):1236-1247.
[32] DIAS A S, ALMEIDA C R, HELGUERO L A, et al. Metabolic crosstalk in the breast cancer microenvironment[J]. Eur J Cancer,2019,121:154-171.
[33] AL-KHAMI A A, ZHENG L Q, DEL VALLE L, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells[J].Oncoimmunology,2017,6(10):e1344804.
[34] MICHALEK R D, GERRIETS V A, JACOBS S R, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+T cell subsets[J]. J Immunol,2011,186(6):3299-3303.
[35] SHARMA P, HU-LIESKOVAN S, WARGO J A, et al. Primary,adaptive, and acquired resistance to cancer immunotherapy[J]. Cell,2017,168(4):707-723.
[36] GELTINK R, KYLE R L, PEARCE E L. Unraveling the complex interplay between T cell metabolism and function[J]. Annu Rev Immunol,2018,36:461-488.
[37] WAICKMAN A T, POWELL J D. mTOR, metabolism, and the regulation of T-cell differentiation and function[J]. Immunol Rev,2012,249(1):43-58.
[38] FRAUWIRTH K A, RILEY J L, HARRIS M H, et al. The CD28 signaling pathway regulates glucose metabolism[J]. Immunity, 2002,16(6):769-777.
[39] HO P C, BIHUNIAK J D, MACINTYRE A N, et al.Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses[J]. Cell,2015,162(6):1217-1228.
[40] DE ROSA V, GALGANI M, PORCELLINI A, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression ofFOXP3exon 2 splicing variants[J]. Nat Immunol,2015,16(11):1174-1184.
[41] HAAS R, SMITH J, ROCHER-ROS V, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions[J]. PLoS Biol, 2015, 13(7):e1002202.
[42] KUMAGAI S, KOYAMA S, ITAHASHI K, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments[J]. Cancer Cell,2022,40(2):201-218.e9.
[43] WATSON M J, VIGNALI P D A, MULLETT S J, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid[J].Nature,2021,591(7851):645-651.
[44] KIDANI Y, ELSAESSER H, HOCK M B, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity[J]. Nat Immunol,2013,14(5):489-499.
[45] YANG W, BAI Y B, XIONG Y, et al. Potentiating the antitumour response of CD8+T cells by modulating cholesterol metabolism[J].Nature,2016,531(7596):651-655.
[46] WANG H P, FRANCO F, TSUI Y C, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors[J]. Nat Immunol,2020,21(3):298-308.
[47] ZENG H,YANG K,CLOER C,et al. mTORC1 couples immune signals and metabolic programming to establish Treg cell function[J]. Nature,2013,499(7459):485-490.
[48] TAKE Y, KOIZUMI S, NAGAHISA A. Prostaglandin E receptor 4 antagonist in cancer immunotherapy: mechanisms of action[J]. Front Immunol,2020,11:324.
[49] MUNN D H, SHARMA M D, BABAN B, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase[J]. Immunity,2005,22(5):633-642.
[50] GEIGER R, RIECKMANN J C, WOLF T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity[J]. Cell,2016,167(3):829-842.e13.
[51] BIAN YJ, LI W, KREMER D M, et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation[J]. Nature,2020,585(7824):277-282.
[52] MOLLER S H, HSUEH P C, YU Y R, et al. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging [J]. Cell Metab,2022,34(3):378-395.
[53] LIU Y N, YANG J F, HUANG D J, et al. Hypoxia induces mitochondrial defect that promotes T cell exhaustion in tumor microenvironment through MYC-regulated pathways[J]. Front Immunol,2020,11:1906.
[54] HE J L, SHANGGUAN X, ZHOU W, et al. Glucose limitation activates AMPK coupled SENP1-Sirt3 signalling in mitochondria for T cell memory development[J]. Nat Commun,2021,12(1):4371.
[55] SCHARPING N E, MENK A V, MORECI R S, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction[J].Immunity,2016,45(2):374-388.
[56] KURAI J, CHIKUMI H, HASHIMOTO K, et al. Antibodydependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines[J]. Clin Cancer Res,2007,13(5):1552-1561.
[57] PITZALIS C, JONES G W, BOMBARDIERI M, et al. Ectopic lymphoid-like structures in infection, cancer and autoimmunity[J].Nat Rev Immunol,2014,14(7):447-462.
[58] CASSIM S, POUYSSEGUR J. Tumor microenvironment: a metabolic player that shapes the immune response[J]. Int J Mol Sci,2019,21(1):157.
[59] WATERS L R, AHSAN F M, WOLF D M, et al. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling[J]. iScience,2018,5:99-109.
[60] BROWN T P, GANAPATHY V. Lactate/GPR81 signaling and proton motive force in cancer: role in angiogenesis, immune escape,nutrition, and Warburg phenomenon[J]. Pharmacol Ther, 2020, 206:107451.
[61] KOUIDHI S, BEN AYED F, BENAMMAR ELGAAIED A.Targeting tumor metabolism: a new challenge to improve immunotherapy[J]. Front Immunol,2018,9:353.
[62] HALFORD S E R,JONES P,WEDGE S,et al. A first-in-human firstin-class (FIC) trial of the monocarboxylate transporter 1 (MCT1)inhibitor AZD3965 in patients with advanced solid tumours[J]. J Clin Oncol,2017,35(15_suppl):2516.
[63] OH M H,SUN I H,ZHAO L,et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells[J]. J Clin Invest,2020,130(7):3865-3884.
[64] LEONE R D, ZHAO L, ENGLERT J M, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion[J]. Science,2019,366(6468):1013-1021.
[65] VOSS K, LUTHERS C R, POHIDA K, et al. Fatty acid synthase contributes to restimulation-induced cell death of human CD4 T cells[J].Front Mol Biosci,2019,6:106.
[66] FALCHOOK G, INFANTE J,ARKENAU H T, et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors[J]. EClinicalMedicine, 2021, 34:100797.
