异噁唑草酮中间体合成技术综述
2022-10-18林子婷耿梅
林子婷,耿梅
(国家知识产权局专利局专利审查协作江苏中心,江苏 苏州 215163)
0 引言
异噁唑草酮,通用名isoxaflutole,CAS RN.141112-29-0,化学名为5-环丙基-4-(2-甲磺酰基-4-三氟甲基)苯甲酰基异噁唑,结构式如图1所示,为罗纳-普朗克公司(现属拜耳公司)开发的对羟基苯基丙酮酸酯双氧化酶抑制剂(HPPD抑制剂),能用于玉米和甘蔗田等旱作物田的防除禾本科杂草和阔叶杂草。因其持效期适中,在土壤中的半衰期比较短,通常在4个月后基本无残留,对后茬作物没有不良影响,在农业生产领域被广泛使用[1]。
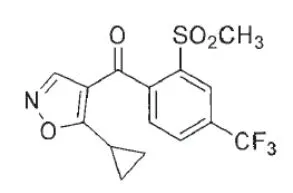
图1 异噁唑草酮结构
根据甲硫基氧化的先后,异噁唑草酮的合成分为前氧化法和后氧化法。前氧化法是指在成环之前先将硫醚氧化成砜基,制备1-环丙基-3-(2-甲磺酰基-4-三氟甲基苯基)丙-1,3-二酮,后氧化法是指先合成环丙基-4-(2-甲硫基-4-三氟甲基)苯甲酰基异噁唑,在工艺最后一步再将甲硫基氧化成砜得到目标产物。无论是前氧化还是后氧化,中间体1-环丙基-3-(2-甲硫基-4-三氟甲基苯基)丙-1,3-二酮都是重要中间体,是重要的化工生产原料之一,因此针对该中间体的合成工艺做详细梳理。
1 2-硫甲基-4-三氟甲基苯甲酯与环丙基甲酸酯缩合反应
2-硫甲基-4-三氟甲基苯甲酯与环丙基甲酸酯在碱性试剂作用下发生Claisen缩合反应得到1-环丙基-3-(2-甲硫基-4-三氟甲基苯基)丙-1,3-二酮。这是经典Claisen缩合反应,CN98805679.8A[2]指出利用酮与酯或碳酸酯的缩合反应效果通常较差,反应收率较低,产品纯度不高,分离较为复杂,可以通过替换溶剂为DMSO来优化反应收率和产品纯度。2-硫甲基-4-三氟甲基苯甲酯与环丙基甲酸酯缩合反应如图2所示。

图2 2-硫甲基-4-三氟甲基苯甲酯与环丙基甲酸酯缩合反应
2 2-硫甲基-4-三氟甲基苯乙酮与环丙烷甲酸酯类缩合制备
CN97191908.9A[3]以2-甲硫基-4-三氟甲基苯乙酮为原料,和环丙烷甲酸酯类物质在甲醇钠等强碱存在的条件下进行缩合反应,收率达到75%。反应式如图3所示。
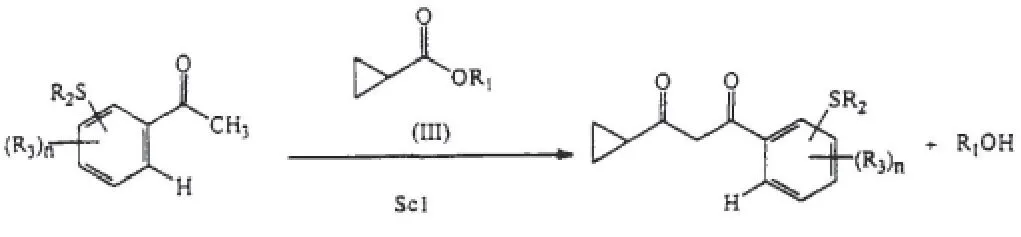
图3 2-硫甲基-4-三氟甲基苯乙酮与环丙烷甲酸酯类缩合制备
CN202010562693.9A[4]专利公开了2-甲硫基-4-三氟甲基苯乙酮的制备路线。包括以对卤三氟甲苯为原料,在混酸中反应引入硝基,利用Heck反应在金属钯和有机膦试剂催化下与乙烯基甲醚反应生成中间体2-硝基-4-三氟甲基苯乙酮,最后与甲硫醇钠反应得到2-甲硫基-4-三氟甲基苯乙酮。第二条路线为利用Nef反应制备2-甲硫基-4-三氟甲基苯乙酮。其首先在氢氧化物的存在下,在极性非质子溶剂中,使具有硝基邻位的卤代的硝基苯化合物与硝基烷烃接触,以产生邻硝基芳烃化合物,然后将邻硝基芳烃化合物氧化以产生邻硝基芳基酮,任选地在催化剂存在下,邻硝基芳基酮与硫醇盐阴离子,例如硫醇钠反应生成邻硫醚2-甲硫基-4-三氟甲基苯乙酮。该路线需要使用易爆品硝基乙烷,生产风险高,不易于工业化。
CN98806963.6A[5]中将 1-溴-2-硝基-4-三氟甲基苯制成格氏试剂,然后引入酮基,最后在甲硫醇钠的作用下发生取代反应生成目标中间体2-甲硫基-4-三氟甲基苯乙酮。格氏反应需要严格无水,反应条件苛刻,后处理工序复杂,三废较高。
苯乙酮类化合物的制备还可以通过取代苯甲腈与碘代烷烃的反应、苯甲酸酯与碘甲烷、三氟甲基苯甲醛和重氮甲烷反应得到。但同样,重氮甲烷依然属于易燃易爆危险品,碘甲烷存在剧毒且容易挥发,上述路线均存在原料获取困难、对生产条件要求高、后处理难度高的缺陷[6]。
3 2-硝基-4-三氟甲基苯甲酯或酸与环丙基甲基酮缩合反应
1-环丙基-3-(2-甲磺酰基-4-三氟甲基苯基)丙-1,3-二酮另一代表性合成路线是以2-硝基-4-三氟甲基苯甲酸为原料,经过甲酯化得到2-硝基-4-三氟甲基苯甲酸甲酯,与甲硫醇钠反应得到2-甲硫基 -4-三氟甲基苯甲酸甲酯,最后与环丙基甲基酮在碱性条件下缩合得到目标化合物。
2-硝基-4-三氟甲基苯甲酯或酸与环丙基甲基酮缩合反应如图4所示。

图4 2-硝基-4-三氟甲基苯甲酯或酸与环丙基甲基酮缩合反应
京博农化科技股份有限公司在2017年3月提出申请CN201710177420.0A[7],以邻甲硫基对三氟甲基苯腈为起始原料,在HCl气体催化作用下与甲醇反应,得到2-甲硫基-4-三氟甲基苯甲酸甲酯,分批加入环丙基甲基酮,最终制备得到1-环丙基-3-(2-甲硫基-4-三氟甲基苯基)丙-1,3-二酮。在酯化步骤中,甲醇既作为反应底物也作为溶剂,降低了反应溶剂的使用量,相较于之前工艺降低了溶剂的用量,最终产品含量可达98%,收率88%。
WO9414782A1[8]、EP0470856A1[9]等 则 以 2-甲硫基-3-三氟甲基苯甲酸为原料,与环丙基甲基酮经由克莱森缩合反应得到1,3-二酮。相较于前述以2-硝基-4-三氟甲基苯甲酸酯为原料进行缩合反应的工序,该工艺省略了一步酯化步骤,精简了反应路线,但反应温度高(300~500 ℃),条件苛刻,需要使用催化床等复杂装置,对生产条件要求较高。
关于2-甲硫基-3-三氟甲基苯甲酸的制备,EP524018A[10]公开了以2-溴-5-(三氟甲基)苯胺为原料,与二甲基二硫醚反应,在烃基锂的作用下通过羰基化试剂CO2引入羧基,最后制备得到2-甲硫基-3-三氟甲基苯甲酸。US5744021A[11]则以3,4-二氯三氟甲基苯为原料进行电化学反应,以CO2为电解质生成2-甲硫基-4-三氟甲基苯甲酸。CN200910087066.8[12]、CN200910232349.7[13]等专利中以4-溴三氟甲基苯为原料,先进行硝基化反应引入硝基,然后在烷基锂作用下引入羧基,得到目标化合物。
针对上述三条路线,牛纪凤[6]指出,原料2-溴-5-(三氟甲基)苯胺和t-BuONO等较难获得,烃基锂试剂保存条件高、反应条件苛刻,有机电解反应又需要特定的装置设备,生产成本高昂,工业化困难。其提出以4-溴-三氟甲基苯为原料,利用混酸进行硝化反应,采用CuCN引入氰基,利用CH3SNa引入甲硫基,得到2-甲硫基-4-三氟甲基苯腈,最后水解得到2-甲硫基-4-三氟甲基苯甲酸。该路线合成步骤虽然较多,共需要4步,但原料易得,反应条件相对温和,操作简便,总收率达到56.5%,HPLC含量98.2%,易于工业化生产。
US5705674A[14]同样公开了以4-三氟甲基-2-硝基卤代苯与氰化物反应制备4-溴-3-硝基三氟甲基苯。诸暨合力化学对外贸易有限公司在2014年12月提出的制备异噁唑类化合物的方法专利CN201410720121.3A[15]中指出,这一反应需使用氰化盐等高毒性原料,且原料2-硝基-4-三氟甲基苯甲酸不易获得。其提出以式I所示的硝基苯类化合物为原料,与RCOCH2CN、氧化剂在-10~60 ℃条件制备2-硝基-4-三氟甲基苯甲酸,其中L为卤素、磺酰基或亚磺酰基。相较于在先工艺,其避免了使用高毒性的氰化物所带来的的安全和三废问题。该公司随后还以该申请为优先权基础提出PCT申请。
US2419259A[16]、US5763627A[17]、CN981111147.5[18]等专利公开了环丙基甲基酮的制备。主要包括以α-烷基酰基-γ-丁内酯为原料,水解开环、脱卤环化生成环丙基甲基酮。或以邻苯二甲胺为起始原料,在PPA催化下,与环丙基甲酸反应生成环丙基苯并咪唑,经由苯并咪唑盐中间体与格氏试剂反应、水解生成环丙基甲基酮和邻苯二甲胺。或以4-羟基-甲基丁基酮为原料,经过卤代、环化反应制得。
US6265618B1[19]中使用催化管式反应器,以羧酸、水、环丙烷甲醛为原料进行气相反应制备不对称酮,实现了环丙基甲基酮的高选择性、高转化率。过使羧酸与铌催化剂在升高的温度下接触来制备酮的方法。该方法使用改性的碱交换铌催化剂,特别适用于由环丙烷羧酸和乙酸生产环丙基甲基酮。
上海创诺提出在以α-乙酰基-γ-丁内酯为原料制备环丙基甲基酮的工艺中,使用混合催化剂可实现一步法制备,所述催化剂包括两组分:(1)NaI、NaBr、KI、KBr;(2)FeI2、ZnI2、CuI2、AgBr、FeBr2、MgBr2、CuBr2、ZnBr2。通过该催化剂,环丙基甲基酮的收率提高至92.5%以上,纯度达到99.3%以上,减少了异构体杂质的生成,显著提高了选择性[20]。
瑞孚信江苏药业以乙酰丙酸甲酯为起始原料,经由3-(2-甲基-1,3-二氧杂环戊烷-2-基)丙酸甲酯、3-(2-甲基-1,3-二氧杂环戊烷-2-基)丙基-1-醇制备5-氯-2-戊酮,最后环化得到环丙基甲基酮。相对于传统工艺使用的乙酰丙醇,乙酰丙酸甲酯更容易获得,中间体无需精制即可作为下一步反应原料直接使用,工艺流程更为简单[21]。
南京理工大学课题组[22]以环丙基乙炔为原料,以[(IPr)AuCl]为催化剂,甲醇和水存在下、110 ℃反应6 h,柱层析分离得到目标化合物,收率达到86%。催化下发生水解反应制备得到环丙基烷基酮,具体参见CN201310482691.9[22]。再此基础上,该课题组进一步提出将[(IPr)AuCl]和AgOTf联用,在溶剂1,4-二氧六环和水中,微波反应仅需要1 h即得到相应的酮,参见CN104557499A[23]。相较于其他方法,该工序显著缩短了时间,且副产物仅有水,实现了绿色化学。
4 2-甲硫基-3-三氟甲基苯乙酮与环丙基甲酰氯缩合反应
环丙基甲酰氯与2-甲硫基-3-三氟甲基苯乙酮缩合反应得到1,3-二酮,参见EP0470856A[8]、CN200980110829.4[24]、EP0487357A1[25]等专利。由于反应底物活性差异,酰氯和酮的缩合反应收率较低,工业化生产效率低。
5 β-氨基乙烯基酮水解制备1,3-二酮
WO95/00476A[26]公开了经由β-氨基乙烯基酮制备 1-环丙基-3-(2-甲硫基-4-三氟甲基苯基)丙-1,3-二酮的方法,其首先利用甲基酮与芳香腈进行缩合反应得到β-氨基乙烯基酮,随后在强无机酸或有机酸存在下进行水解得到1,3-二酮。以该方法制备中间体β-氨基乙烯基酮时,其会生成少量嘧啶或酰胺杂质,以摩尔%表示,基于氨基乙烯酮的量,会存在不超过14%的嘧啶和35%的酰胺。利用甲苯或甲苯/环己烷对产物进行重结晶可以得到纯的β-氨基乙烯基酮。
江苏中旗科技股份有限公司在申请CN201911326745.6[27]中指出以 2-(甲硫基)-4-(三氟甲基)苯甲酸甲酯和环丙基甲基酮为原料发生克莱森缩合反应的方法对反应水分含量有严格要求,极易水解生成2-(甲硫基)-4-三氟甲基苯甲酸,原料转化率降低,环丙基甲基酮自身发生羟醛缩合进一步消耗了原料,生成难以分离的高聚物杂质。已有的工艺反应温度高,选择性和收率差。以2-(甲硫基)-4(三氟甲基)苄腈和环丙基甲基酮反应,在制备氨基乙烯酮的过程中,含有高于14%的嘧啶和酰胺杂质,产品纯度难以控制,收率低,产物精制困难。CN113004179A中以1-(2-(甲硫基)-4-(三氟甲基)苯基)乙酮和环丙烷甲腈为起始原料在30~100 ℃下发生缩合反应得到中间体 3-氨基-3-环丙基-1-(2-(甲硫基)-4-(三氟甲基)苯基)-2-烯-1-丙酮,该亚胺化合物在强酸条件下发生水解最终得到1-环丙基-3-(2-甲硫基-4-三氟甲基苯基)丙-1,3-二酮,两步反应累计收率达到97%以上,显著提高了收率,避免了嘧啶和酰胺等杂质的生成。
6 结语
通过对1-环丙基-3-(2-甲硫基-4-三氟甲基苯基)丙-1,3-二酮的合成技术进行梳理,为今后国内化工生产企业的研发人员提供合适的知识储备,为今后开发更好的合成方法寻找合适的突破方向,进而推动技术革新。
