甘草次酸固体分散体3种制备工艺比较*
2022-10-12刘婷婷隋小宇
潘 虹,李 飞,柴 硕,刘婷婷,刘 畅,隋小宇
(齐齐哈尔医学院药学院,黑龙江 齐齐哈尔 161006)
甘草次酸(GA)是中药甘草主要活性成分甘草酸的水解产物[1],属五环三萜化合物,易溶于甲醇、乙醇等有机溶剂,热稳定好,具有抗炎、抗感染、抗肿瘤、镇咳祛痰、免疫抑制作用,临床多用于慢性肝炎和肝癌的治疗[2-6]。但GA的水溶性较差,口服生物利用度低,限制了其临床应用[7]。目前多通过制备微乳、纳米粒、环糊精包合物、固体分散体、脂质体等剂型加以改善[8]。固体分散(SD)技术可将药物以分子、微晶、非晶态的形式均匀分散在固体载体中,是提高低溶性药物的溶出度和口服生物利用度的有效方法[9-11]。固体分散体常用的制备方法有熔融法、溶剂法、溶剂-熔融法、喷雾(冷冻)干燥法和研磨法,其中前三者为目前应用较多、且较易实现的方法。熔融法具有时间短、无溶剂、成本低的优点,溶剂法具有操作简便、药物分散均匀、无需高温的优点,溶剂-熔融法的温度和混合时间均小于熔融法,可使药物免受热降解。此外,与溶剂法相比,熔融状态的载体更易分散和溶解[12-13]。近年来,超临界流体技术、喷雾(冷冻)干燥技术、热熔挤出技术等新型技术也在逐步发展[14-15]。聚乙二醇(PEG)是由环氧乙烷聚合得到的水溶性聚合物,易溶于水和多种有机溶剂,在热、酸、碱条件下化学性质稳定,熔点低、固化速度快,具有良好的生物相容性,活性较低,且无毒无刺激性[16-18],适宜作为上述3种方法制备固体分散体时的载体。本研究中以PEG4000为载体,GA为主药,比较上述3种方法制备的固体分散体的溶出性质和理化性质,为GA口服制剂的开发提供参考。现报道如下。
1 材料与方法
1.1 仪器与试药
仪器:LC-1000型高效液相色谱仪(山东鲁南瑞虹化工仪器有限公司);RCZ-8AⅡ型自动溶出仪(天津大学精密仪器厂);S-4300型扫描电子显微镜(日本Hitachi公司);Nicolet 6700型傅里叶红外光谱仪(赛默飞世尔科技<中国>有限公司);HSC-1型DSC差示扫描量热仪(北京恒久实验设备有限公司);D8 Advance型聚焦X射线衍射仪(德国Bruker公司)。
试药:GA原料药(大连美仑生物技术有限公司,批号为471-53-4,含量≥98%);GA对照品(中国食品药品检定研究院,批号为110723-201514,含量≥99%);PEG4000、无水乙醇(国药集团化学试剂有限公司)。
1.2 方法
1.2.1 样品(GA固体分散体)制备
熔融法:将处方量的GA原料药及PEG4000置蒸发皿中,于70~80℃水浴加热至熔融,搅拌。待药物颗粒消失后,倒于不锈钢板上形成薄层,于-20℃迅速冷却固化2 h,取出后在干燥器内干燥数天,研磨粉碎,过80目筛,得样品1,备用。
溶剂-熔融法:将处方量的PEG4000置蒸发皿中,于70~80℃水浴加热至熔融。将GA原料药溶解于少量无水乙醇中,与熔融的PEG4000混合均匀,待无水乙醇完全挥发后,倒于不锈钢板上形成薄层,于-20℃迅速冷却固化2 h,取出后在干燥器内干燥数天,研磨粉碎,过80目筛,得样品2,备用。
溶剂法:将处方量的GA原料药及PEG4000置蒸发皿中,于80~90℃水浴加热溶解于无水乙醇中,搅拌,待溶剂完全挥发后,置干燥器内干燥,研磨粉碎,过80目筛,得样品3,备用。
1.2.2 体外溶出度考察
参考2020年版《中国药典(二部)》附录桨法,以900 mL磷酸盐缓冲液(PBS,pH 6.8)作为固体分散体(相当于10.0 mg药物)的溶出介质,按药载比1∶2,1∶4,1∶6,1∶8取样品粉末适量,精密称定,于(37±0.5)℃下,100 r/min转动5,15,30,45,60,75,90,120 min时分别吸取5 mL,经0.45 μm醋酸纤维素膜滤过,取续滤液,同时补充等体积同温介质5 mL。采用高效液相色谱法测定释药量,并计算GA累积溶出度。色谱条件,色谱柱为Zorbax C18柱(150 mm×4.6 mm,5 μm),流动相为甲醇-水-冰醋酸(88∶11∶1,V/V/V),续滤液超声脱气,流速为1.0 mL/min,检测波长为250 nm,柱温为室温,进样量为20 μL[19]。以峰面积(A)为纵坐标、质量浓度(C,μg/mL)为横坐标进行线性回归,得回归方程A=0.478C+0.080 7(R2=0.999 9),GA质量浓度在2.54~101.60 μg/mL范围内与峰面积线性关系良好。方法学考察试验结果的RSD均小于2.0%,表明精密度、溶液稳定性、方法重复性均良好。
1.2.3 结构表征
差示扫描量热(DSC)分析:采用DSC扫描量热仪。参比物为空铝坩埚,扫描范围为25~350℃,升温速率为10℃/min,保温时间为10 min。药载比为1∶8(m/m),下同。
X-射线衍射(XRD)分析:采用聚焦X射线衍射仪。分析条件为,Cu-Kα靶,最大电压为40 kV,电流为40 mA,扫描范围为5°~50°,扫描速率为5°/min。
傅里叶变换红外光谱(FTIR):采用傅里叶红外光谱仪。将样品与溴化钾在玛瑙研钵中混合,压片。扫描范围为4 000~400 cm-1,分辨率为4 cm-1。
傅里叶变换拉曼光谱(FTIR-Raman):采用装有NXR FT-Raman模块(1 064 nm)的傅里叶红外光谱仪。扫描范围为3500~500 cm-1,累计扫描64次,分辨率为4 cm-1,光源设置外部光,CaF2分束镜。
扫描电子显微镜(SEM)分析:采用扫描电子显微镜。取少量样品粉末,均匀黏附于导电胶,喷金处理后,置样品台上抽真空观察。分析条件为,真空镀金5 min,加速压力20.0 kV。
2 结果
2.1 溶出曲线比较
不同制备方法对溶出度有明显影响,从曲线斜率看,当药载比相同时,溶出度的大小顺序为溶剂法>溶剂-熔融法>熔融法。药载比对溶出度也有明显影响,随着药载比从1∶2(m/m)变至1∶8(m/m),样品的溶出度均逐渐增大,并且明显高于GA原料药的溶出度。在120 min时,3种方法制备样品的累积溶出度不同,药载比为1∶8(m/m)时,通过溶剂法、溶剂-熔融法和熔融法制备的样品累积溶出度分别为94.17%,88.17%,77.89%。综上,溶剂法制备的样品具有更优的溶出度。详见图1。
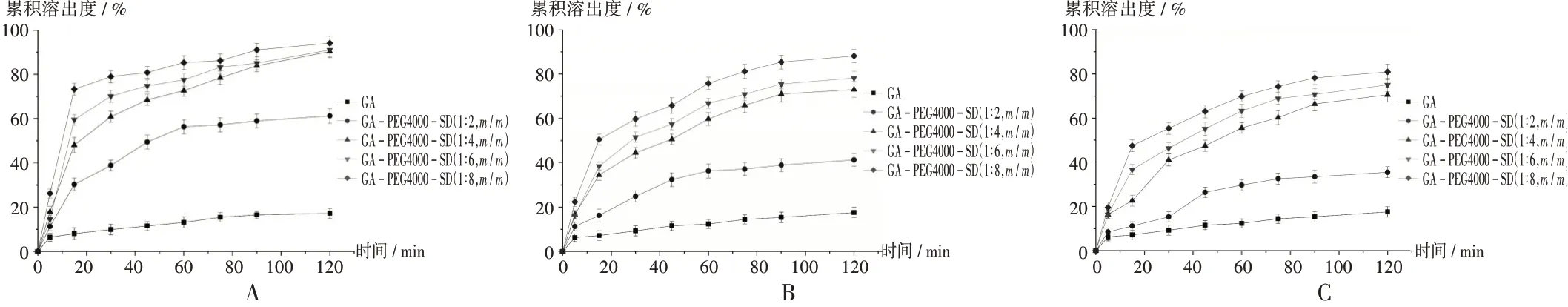
图1 溶出曲线A.Solvent method B.Solvent-melting method C.Melting methodFig.1 Profile of dissolution rate
2.2 DSC比较
热分析技术可用于推测药物结晶度或药物-载体相互作用的变化,若药物发生非定形或与载体相互作用,相应的熔点峰就会消失或减弱[20-21]。DSC谱图见图2。可见,GA在301℃处有明显的熔融吸热峰,即GA原料药的熔点为301℃;PEG4000在60℃和288℃处均有熔融吸热峰,其中60℃处峰较尖锐,288℃处峰较宽;物理混合物(PM)在280℃处有尖锐的吸热峰,为PEG4000和GA在此处吸热效应叠加的结果;样品1,2,3均在250℃之后未出现尖锐的吸热峰,而分别在291,290,286℃处开始出现非常平缓的吸热峰。从DSC谱图变化可推断,固体分散体形成后,GA的晶体结构转变为微晶,且熔点发生偏移,且样品3的熔点偏移更大,表明其结晶度降低得更明显。

图2 差示扫描量热分析谱图a.GA b.PEG4000 c.PM d.Sample 1 e.Sample 2 f.Sample 3Fig.2 DSC spectra
2.3 XRD比较
结晶性药物在不同的衍射带中均具有晶体的特征衍射峰,这些峰在形成固体分散体后消失或减弱,表明药物以非晶态或微晶形态存在于固体分散体中[22]。XRD谱图见图3。可见,GA在5°~16°之间有6个明显的特征衍射峰;PEG4000在19°和23°处有2个特征衍射峰;PM的XRD谱图中可观察到GA和PEG4000各自的特征衍射峰;样品1,2,3的XRD谱图中,均可见到较强的PEG4000两处特征衍射峰,但GA的特征衍射峰(15°处左右)的平均峰高均降低,其中样品2,3特征衍射峰平均峰高均低于样品1;与样品2相比,样品3的特征衍射峰平均峰高较低。综上,溶剂法制备样品的结晶度明显低于其他两种方法,且3种样品的溶出度与其结晶度呈负相关。
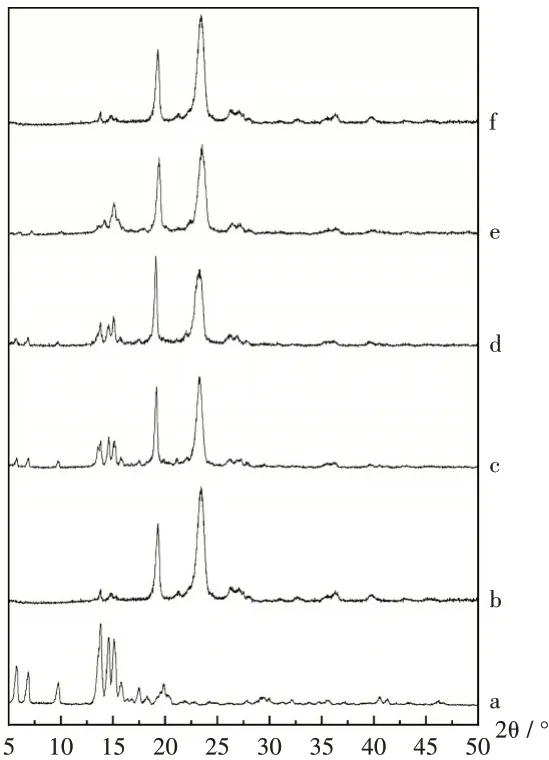
图3 X-射线衍射分析谱图a.GA b.PEG4000 c.PM d.Sample 1 e.Sample 2 f.Sample 3Fig.3 XRD patterns
2.4 FTIR比较
固体分散体可增加与药物-载体相互作用相关的药物溶解度,药物与载体之间的氢键对于解释药物在固体分散体中的物理状态和稳定性非常重要[23]。本研究中将FTIR用于检测固体分散体系潜在的分子间相互作用。FTIR图谱见图4。GA在3 444,1 713,1 663 cm-1处有3个特征吸收峰,分别为GA的3号位—OH、11号位—CO—以及30号位—COOH的伸缩振动峰;PEG4000在3 448 cm-1和2 887cm-1处有2个特征吸收峰,分别为—OH的伸缩振动峰和—CH2—的伸缩振动峰;样品1,2,3的红外光谱中,GA的3号位—OH的吸收峰(3 444 cm-1)分别出现在3 441,3 442,3 440 cm-1处,且其峰宽较GA的峰更宽。可能是GA与PEG4000形成氢键而缔合的—OH伸缩振动成一系列多重叠峰,或是与PEG4000的—OH伸缩振动相叠加所致;样品1,2,3的FTIR光谱中,GA原料药的—CO—在1 713 cm-1处的特征吸收峰也分别移至1 705,1 703,1 704 cm-1处。这种向较低波长的转变可能是由于GA上C3的—OH或环上的—CO—与PEG的ROH之间形成了分子间氢键。

图4 红外光谱图a.GA b.PEG4000 c.PM d.Sample 1 e.Sample 2 f.Sample 3Fig.4 Infrared spectras
2.5 FTIR-Raman比较
拉曼光谱可用于固体分散体中聚合物与药物的相互作用分析[24]。拉曼光谱见图5。可见,GA在1 665 cm-1拉曼位移处有2个尖锐且可与PEG4000进行明显区分的特征峰,并且GA及PEG4000在2 880 cm-1附近各有一个较大的散射峰。因此,选取这2处特征峰与3种样品拉曼光谱图比较,由于分子间相互作用行为不同,非晶态和晶体态的拉曼光谱也有所不同。与晶体相比,非晶体形式的拉曼光谱有更宽的峰,由此反映了非晶形的随机状态[25]。2 880 cm-1处半峰宽大小顺序为样品3>样品2>样品1>GA,与GA拉曼光谱图相比,3种样品拉曼光谱的半峰宽均有所变大,这可能是固体分散体中GA的晶化率降低所致,且半峰宽的大小顺序与3种样品的溶出度呈正相关。而3种样品在1 665 cm-1处的2个尖锐峰的峰高均较PM低。
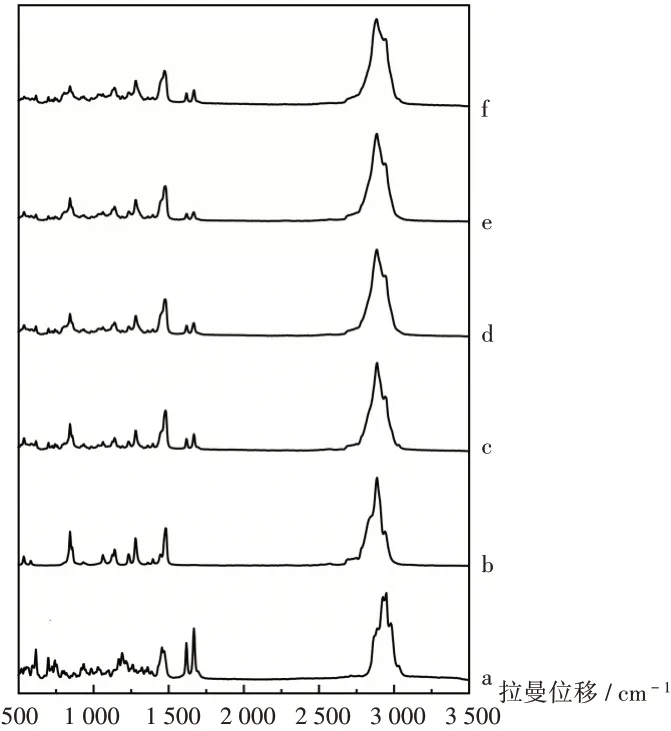
图5 拉曼光谱图a.GA b.PEG4000 c.PM d.Sample 1 e.Sample 2 f.Sample 3Fig.5 Raman micrographs
2.6 SEM比较
SEM图见图6。GA原料药为条状晶体;PEG4000为片状不规则态;3种样品的SEM图无明显差别,均为不规则的片状体,与PEG4000的形态一致,且3种样品的SEM图中未发现GA的条状晶体形态存在。

图6 扫描电子显微镜图a.GA(×20 000)b.PEG4000(×5 000)c.PM(×20 000)d.Sample 1(×10 000)e.Sample 2(×10 000)f.Sample 3(×20 000)Fig.6 SEM graphs
3 讨论
GA难溶于水,口服吸收较差,而将其制备成固体分散体可以改善其溶出度。本研究中采用熔融法、溶剂-熔融法和溶剂法分别制备,结果显示,3种样品的溶出度均明显高于GA原料药,且在相同药载比条件下,样品3的溶出度优于样品1和样品2。从XRD分析可知,样品3的结晶度明显低于样品1和样品2,其抑制GA结晶的能力更强,更易于形成过饱和溶液,从而提高溶出度。另一方面,GA溶出度也与药物的分散程度和粒径尺寸有关,根据Noyes-Whitney方程,随着药物粒子逐渐变小,其比表面积变大,从而使溶出度增加[26],采用溶剂法制备,药物和载体以分子态充分混合,并且以共沉淀析出,混合体系较其他2种样品更均匀。所以,样品3中GA的分散度较其他2种样品更高,药物粒子的比表面积更大,因此具有更好的溶出效果。
本研究存在一定局限性。首先,由于实验室条件有限,仅对上述3种方法进行了比较,一些新颖的方法,如超临界流体技术、熔融挤出法等并未纳入比较;其次,仅选用了PEG作为载体,未考察其他适用的载体;再次,仅对GA的体外溶出度进行了比较,未考察体内溶出度。
目前,固体分散体是解决水溶性不良药物口服生物利用度低问题的有效方法,发展至今已有4代,前3代旨在改善药物的溶出度,而第4代则属于控释胶囊分散体。尽管一些与不稳定性和可伸缩性有关的问题仍存在,但随着学术和工业研究的发展,具有巨大潜力的新颖和优化的制造技术正在引入。对药物和载体的物理化学性质和相互作用的深入研究、不同制备方法对药物溶出的影响以及表征技术的改进可能有助于全面阐明固体分散体的结构和溶解机理。可为难溶性药物提供更好的载体选择和制备方法,从而克服药物的生物利用度问题以及固体分散体的稳定性问题。
