CTRP9对远端缺血预处理心肌梗死模型大鼠心肌缺血再灌注致脑损伤的保护作用及其机制研究*
2022-10-08谢骏高凤敏符北京
谢骏,高凤敏,符北京
(牡丹江医学院附属红旗医院心内科,黑龙江牡丹江 157011)
缺血性心肌病居世界致死原因的首位,具有高发病率和病死率等特点,而缺血再灌注与其病死率居高不下密切相关[1-2]。缺血再灌注损伤主要指血流灌注逐渐恢复后,组织结构受损或功能丧失进一步加重的现象[3-4]。缺血再灌注后造成的损伤较先前更为严重,且持续时间更长,可直接影响患者的重要脏器,致其结构和功能受损。尽早、尽快恢复冠状动脉的血流灌注是治疗心肌梗死,提高患者存活率的关键。MURRY 等[5]提出,心肌缺血预处理可对心肌起到保护作用。有研究证实,吸入麻醉药预处理和后处理可有效减轻心肌缺血再灌注损伤。近年来有研究显示,AMPK 在调节人体氧化应激和细胞凋亡、增殖等过程中发挥了较大作用[6]。激活后的AMPK 可通过多种途径促进ATP 的生成和脂肪酸的氧化。现有临床证据显示,激活状态的AMPK可对心肌细胞起到保护作用[7-8]。CTRP9 是一种新发现的脂肪组织源性细胞因子,参与吞噬细胞清除凋亡细胞、调控机体炎症反应及免疫耐受等生理过程[9]。
目前关于CTRP9 调控AMPK 途径参与远端缺血预处理对心肌梗死模型心肌缺血再灌注致脑损伤的保护作用的报道较少。因此本研究通过复制缺血再灌注模型大鼠,分别给予后处理和CTRP9 抑制剂干预来明确CTRP9 通过调控AMPK 途径发挥心肌保护作用的机制,现报道如下。
1 材料与方法
1.1 材料
1.1.1 实验动物雄性SD 大鼠40 只,SPF 级,体重220~250 g,购自辽宁长生生物技术股份有限公司,实验动物生产许可证号:SCXK(辽)2020-0001,实验动物使用许可证号:SYXK(黑)2021-0002。
1.1.2 主要试剂戊巴比妥钠、苏木精、CTRP9 抑制剂LiCI(美国Sigma 公司),TUNRL 试剂盒(美国罗氏公司),p-AMPK(1∶1 000,批号:2535,美国Cell Signaling 公司)、一抗LC3(1∶1 000,批号:136F3,美国Santa Cruz 公司)、一抗P62(1∶1 000,批号:8G10,美国Santa Cruz 公司)、内参GAPDH(1∶1 000,批号:AG019,上海碧云天生物技术有限公司),苦味酸、伊红Y、二甲苯、石蜡、中性树胶(北京中国国药集团),拘橡酸、拘橡酸钠(北京华科盛精细化工产品贸易公司),链霉菌抗生素蛋白-过氧化物酶、SABC 试剂盒、DAB 显色试剂盒、白细胞介素-6(Interleukin-6,IL-6)多克隆抗体、IL-8 多克隆抗体、IL-10 多克隆抗体(武汉博士德生物工程有限公司),Bal-2 多克隆抗体、Bax 多克隆抗体(北京中杉金桥生物技术有限公司)。
1.1.3 主要仪器高精度电子天平、pH 计(PB10)(德国Sartorius 公司),移液器、离心机(德国Eppendorf 公司),切片机(德国Leica 公司),PVDF 膜(美国Millipore 公司),荧光显微镜(德国Axiophot 公司),全自动生化分析仪(美国Beckman 公司),Image-Pro6.0 计算机图像分析软件(美国Media Cybemetics 公司),凝胶成像系统、电泳槽(美国Bio-Rad 公司),普通光学显微镜、BX50 显微摄影系统(日本Olympus 公司),V30 扫描仪(日本Epson 公司),-70℃深低温冰箱(日本SANYO 公司),Bene View T5 型监护仪(中国西安巨田医疗设备有限公司),动物呼吸机(上海嘉鹏科技有限公司),微型漩涡混合仪(上海沪西分析仪器有限公司),TGL-16G型高速台式离心机(上海医用电子仪器厂)。
1.2 方法
1.2.1 动物模型复制及分组所有大鼠插管开胸后,复制心肌缺血再灌注模型前,置于手术台约10 min,期间不做任何处理,便于大鼠适应呼吸机、开胸操作的损伤及胸腔压力的改变。实验期间对动物的处理均严格按照相关伦理学规定进行。将大鼠按随机数字表法分为4 组,每组10 只:①假手术组(NS组):大鼠在左心耳下缘位置心肌组织穿线,但对冠状动脉左前降支不做结扎。②缺血再灌注组(NIR组):大鼠在左心耳下缘位置穿线结扎冠状动脉左前降支,进行缺血再灌注处理,缺血时长30 min,松开结扎线后继续灌注2 h。③缺血预处理组(NIPost组):大鼠在左心耳下缘位置穿线结扎冠状动脉左前降支,缺血时长30 min,松开结扎线后再行3 次处理(将结扎线放松再灌注10 s,拉紧结扎线缺血10 s,循环3 个周期,最后松开结扎线行2 h 再灌注)。④缺血预处理+CTRP9 抑制剂组(NIPostI 组),术前10 min灌注0.3 mmol/kg 0.5% LiCl 抑制剂,余下后处理操作方法与NIPsot 组一致。
1.2.2 标本采集再灌注120 min 后迅速取脑,4℃、PBS 溶液清洗,去除残余血液,取适量脑组织置于-80℃冰箱备用。用10%甲醛溶液固定剩余脑组织,用于制作组织切片。
1.2.3 苏木精-伊红(hematoxylin-eosin, HE)染色观察大鼠脑组织病理学变化取适量脑组织,梯度乙醇脱水处理,透明剂(二甲苯)处理后包埋至蜡块中,冷冻后切成约4 μm 厚切片。组织脱蜡、水化后,苏木精染色5~8 min,70%乙醇1 min,2%碳酸氢钠返蓝5~10 s,二甲苯透明处理2 min,最后以中性树胶封片。封片后置于光镜下观察脑组织病理学变化。
1.2.4 免疫组织化学染色检测大鼠脑组织凋亡因子、炎症因子取1.2.3 中组织切片置于40℃水浴锅中摊片,60℃烤箱烤片3 h 后脱蜡处理。采用微波对脱蜡水化的组织切片进行抗原修复,滴加3%过氧化氢溶液,以阻断内源性过氧化物酶的影响。使用免疫组织化学笔在组织周围画圈,滴加合适浓度的山羊血清予以封闭。依次加一抗、酶标二抗、显色剂,苏木精复染后脱水、风干后切片,以中性树胶进行封片,并于显微镜下观察切片并拍照。结果判定标准:相同光强度下,400 倍显微镜下各个切片随机挑选5 个互不重叠的视野,细胞内有棕黄色颗粒即为阳性。
1.2.5 TUNEL 法检测大鼠脑组织细胞凋亡取1.2.3 中组织切片贴附在多聚赖氨酸预处理后的载玻片上,确保切片的平整且不重叠。将切片置于62℃温箱中烤片12 h,组织切片脱蜡、水化,二甲苯溶液冲洗玻片2 次,3 min/次;无水乙醇冲洗玻片2 次,3 min/次;95%和75%乙醇溶液分别清洗1 次,3 min/次;蒸馏水清洗2 次,5 min/次;PBS 洗2 次,5 min/次;37℃条件下20 μg/mL 蛋白酶K 溶液消化15 min,去除组织蛋白;蒸馏水冲洗4 次,2 min/次。将含2%过氧化氢的PBS 溶液加入色缸中,室温条件下反应5 min,PBS 冲洗2 次,5 min/次,使用滤纸将组织周围水分吸干后,即刻在切片上滴加约50 μL TUNEL 反应液,置于37℃湿盒中孵育1 h。PBS 液冲洗3 次,3 min/次,将切片周围水分甩干后,滴加50 μL 辣根过氧化酶抗体至切片上,置于37℃温箱中孵育30 min。苏木精复染,PBS 液冲洗3 次,3 min/次,甩干水分,将2 滴DAB 溶液滴加在各组织切片上,室温条件下显色3~6 min。PBS 液冲洗3 次,3 min/次,二甲苯脱水,以中性树胶封片、干燥。结果判定标准:细胞核呈棕色为细胞凋亡阳性;细胞核染蓝色为未发生凋亡。在400 倍显微镜下,通过双盲法对每个视野中的阳性细胞进行计数。
1.2.6 Western blotting检测大鼠心肌组织p-AMPK/t-AMPK、LC3Ⅰ/Ⅱ及P62蛋白的表达将大鼠左心室心尖部组织约300 mg 剪碎后置于匀浆机,依次加入裂解液、蛋白酶及磷酸酶抑制剂,低温条件下,在超声细胞破碎仪中进行3 次裂解,裂解后静置30 min,移入EP 管进行离心,将上清液分装至1.5 mL离心管中,保存于低温冰箱。上述所有操作需在冰面进行,用BCA 蛋白定量试剂盒检测蛋白浓度,并将其稀释至相同浓度。将缓冲液倒入样品中并完全混匀,95℃煮5 min,待蛋白质全部变性后转移至低温冰箱。采用BCA 试剂盒检测蛋白样品含量,并通过裂解液将其调至相同浓度,在97℃金属浴条件下持续加热5 min。每个样品提取20 μg 蛋白置于12%聚丙烯酰胺凝胶中电泳,分离蛋白。电泳结束后,切取目的条带,并将其转移至PVDF 膜上。5%脱脂奶粉封闭2 h,分别加入一抗p-AMPK、一抗LC3、一抗P62、内参GAPDH,4℃一抗孵育过夜。TBST 溶液漂洗3 次后加入二抗摇床,孵育120 min。采用ECL 试剂盒进行显色,然后进行增强化学发光反应,将工作液滴加在PVDF 膜上,反应5 min 左右待出现明显的荧光带,用滤纸吸去剩下的底物液,盖上保鲜膜,X 射线胶片压片后分别放入显影液、定影液,放置适宜时间后冲洗胶片。将胶片晾干,扫描,使用Image 软件测定条带灰度值,以目的条带灰度值与GAPDH 灰度值比值表示蛋白相对表达量。
1.3 统计学方法
数据分析采用SPSS 20.0 统计软件。计量资料以均数±标准差(±s)表示,比较用单因素方差分析,进一步两两比较用LSD-t检验,P <0.05 为差异有统计学意义。
2 结果
2.1 各组大鼠脑组织病理学变化
HE 染色结果显示,NS 组皮层神经元胞浆丰富,核圆,碱染后呈蓝色,分布均匀且排列整齐;NIR 组神经元结构受损,分布不均匀,胞浆空泡化,胞核固缩,碱染后呈淡红色;NIPost 组细胞结构基本恢复正常,大部分神经元包膜完整,胞核清晰可见;NIPostI 组经CTRP9 抑制剂LiCl 干预后,后处理的保护效应消失。见图1。

图1 各组大鼠脑组织病理切片 (HE染色×400)
2.2 各组大鼠脑组织凋亡因子表达水平比较
NS 组、NIR 组、NIPost 组、NIPostI 组Bax 和Bcl-2表达水平比较,经方差分析,差异有统计学意义(F=2.368 和2.135,P=0.003 和0.005)。进一步两两比较结果:NIR 组、NIPost 组及NIPostI 组Bax 表达水平高于NS 组(P<0.05),Bcl-2 表达水平低于NS 组(P<0.05);NIPost 组较NIR 组Bax 表达水平降低(P<0.05),Bcl-2 表达水平升高(P<0.05);NIPostI 组较NIPost 组Bax 表达水平升高(P<0.05),Bcl-2 表达水平降低(P<0.05)。见表1。
表1 各组大鼠脑组织Bax和Bcl-2表达水平比较(n=10,±s)
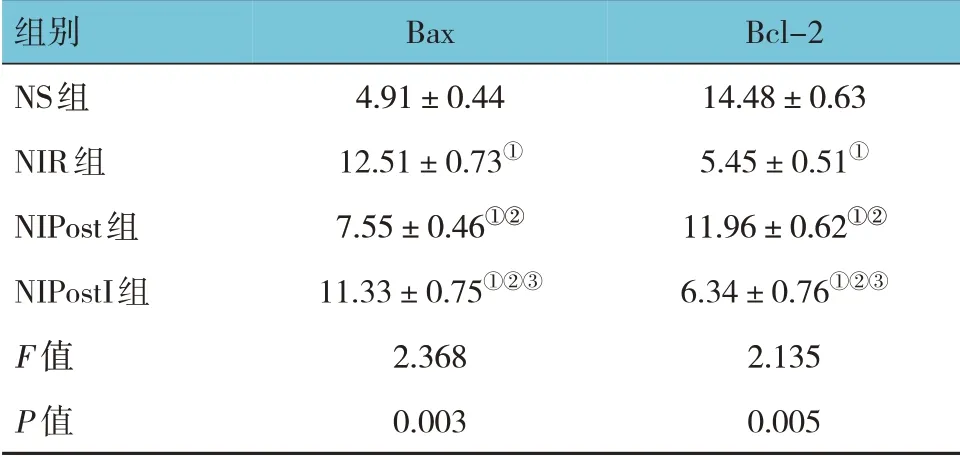
表1 各组大鼠脑组织Bax和Bcl-2表达水平比较(n=10,±s)
注:①与NS 组比较,P <0.05;②与NIR 组比较,P <0.05;③与NIPost组比较,P <0.05。
Bcl-2 14.48±0.63 5.45±0.51①11.96±0.62①②6.34±0.76①②③2.135 0.005组别NS组NIR组NIPost组NIPostI组F 值P 值Bax 4.91±0.44 12.51±0.73①7.55±0.46①②11.33±0.75①②③2.368 0.003
2.3 各组大鼠脑组织炎症因子表达水平比较
NS 组、NIR 组、NIPost 组、NIPostI 组IL-6、IL-8 和IL-10表达水平比较,经方差分析,差异有统计学意义(F=2.123、2.671 和2.935,P=0.011、0.002 和0.001)。进一步两两比较结果:NIR 组、NIPost 组及NIPostI 组IL-6、IL-8表达水平高于NS组(P<0.05),IL-10表达水平低于NS 组(P<0.05);NIPost 组较NIR 组IL-6、IL-8表达水平降低(P<0.05),IL-10 表达水平升高(P<0.05);NIPostI 组较NIPost 组IL-6、IL-8 表达水平升高(P<0.05),IL-10表达水平降低(P<0.05)。见表2。
表2 各组大鼠脑组织IL-6、IL-8及IL-10表达水平比较(n=10,±s)
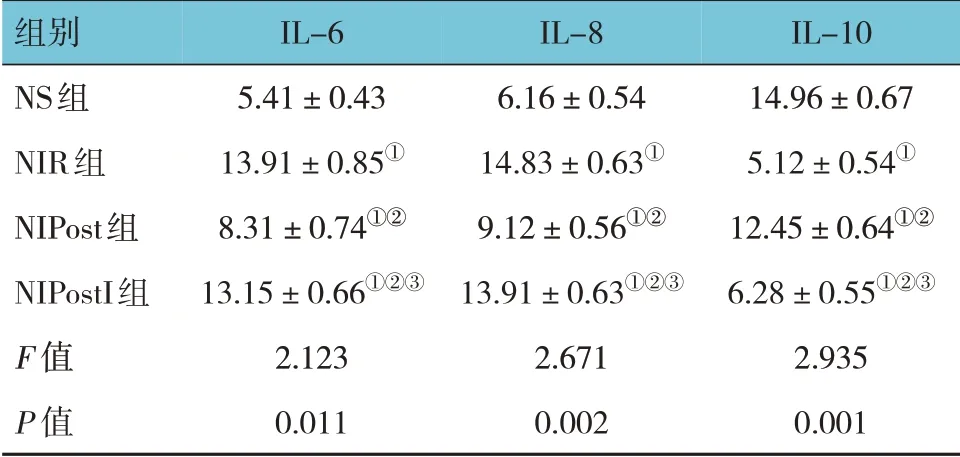
表2 各组大鼠脑组织IL-6、IL-8及IL-10表达水平比较(n=10,±s)
注:①与NS 组比较,P <0.05;②与NIR 组比较,P <0.05;③与NIPost组比较,P <0.05。
IL-10 14.96±0.67 5.12±0.54①12.45±0.64①②6.28±0.55①②③2.935 0.001组别NS组NIR组NIPost组NIPostI组F 值P 值IL-6 5.41±0.43 13.91±0.85①8.31±0.74①②13.15±0.66①②③2.123 0.011 IL-8 6.16±0.54 14.83±0.63①9.12±0.56①②13.91±0.63①②③2.671 0.002
2.4 各组大鼠脑组织细胞凋亡情况
NS 组、NIR 组、NIPost 组、NIPostI 组凋亡细胞数分别为(55.41±6.43)个/HP、(213.91±20.85)个/HP、(138.31±15.74)个/HP、(163.15±10.66)个/HP,经方差分析,差异有统计学意义(F=2.813,P=0.001)。进一步两两比较结果:NIR 组、NIPost 组和NIPostI 组凋亡细胞多于NS 组(P<0.05);NIPost 组和NIPostI 组较NIR 组凋亡细胞减少(P<0.05)。NIPost 组和NIPostI组比较,差异无统计学意义(P>0.05)。见图2。
图2 各组大鼠脑组织细胞凋亡情况 (TUNEL染色×400)
2.5 各组大鼠心肌组织p-AMPK/t-AMPK、LC3Ⅰ/Ⅱ及P62蛋白相对表达量比较
NS 组、NIR 组、NIPost 组、NIPostI 组p-AMPK/t-AMPK、LC3Ⅰ/Ⅱ及P62 蛋白相对表达量比较,经方差分析,差异有统计学意义(F=2.203、2.772 和2.901,P=0.004、0.002 和0.001)。进一步两两比较结果:NIR 组、NIPost 组及NIPostI 组p-AMPK/t-AMPK蛋白相对表达量低于NS 组(P<0.05),LC3Ⅰ/Ⅱ和P62 蛋白相对表达量高于NS 组(P<0.05);NIPost 组较NIR 组p-AMPK/t-AMPK 蛋白相对表达量升高(P<0.05),LC3Ⅰ/Ⅱ和P62 蛋白相对表达量降低(P<0.05);NIPostI 组较NIPost 组p-AMPK/t-AMPK 蛋白相对表达量降低(P<0.05),LC3Ⅰ/Ⅱ和P62 蛋白相对表达量升高(P<0.05)。见表3 和图3~5。

图3 各组大鼠心肌组织p-AMPK/t-AMPK蛋白的表达
表3 各组大鼠心肌组织p-AMPK/t-AMPK、LC3Ⅰ/Ⅱ及P62蛋白相对表达量比较 (n=10,±s)
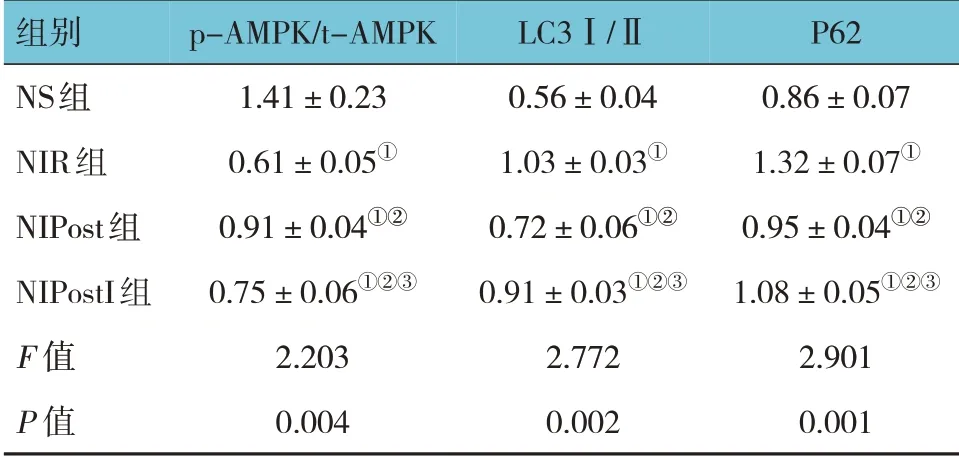
表3 各组大鼠心肌组织p-AMPK/t-AMPK、LC3Ⅰ/Ⅱ及P62蛋白相对表达量比较 (n=10,±s)
注:①与NS 组比较,P <0.05;②与NIR 组比较,P <0.05;③与NIPost组比较,P <0.05。
P62 0.86±0.07 1.32±0.07①0.95±0.04①②1.08±0.05①②③2.901 0.001组别NS组NIR组NIPost组NIPostI组F 值P 值p-AMPK/t-AMPK 1.41±0.23 0.61±0.05①0.91±0.04①②0.75±0.06①②③2.203 0.004 LC3Ⅰ/Ⅱ0.56±0.04 1.03±0.03①0.72±0.06①②0.91±0.03①②③2.772 0.002

图4 各组大鼠心肌组织LC3Ⅰ/Ⅱ蛋白的表达

图5 各组大鼠心肌组织P62蛋白的表达
3 讨论
临床上,及时恢复患者的氧合和血流灌注是抢救缺血再灌注损伤的基础。一旦脏器发生缺血再灌注,所造成的损害将较先前更严重,且更持久,甚至可直接危及心、肺、脑等重要脏器。有研究结果显示,缺血再灌注损伤与氧自由基产生、促凋亡因子过表达、细胞内钙超载及炎症反应的级联效应等密切相关,但具体机制尚未明确[10-11]。1986年,MURRY等[5]首次观察到心肌在经历一次或反复数次短暂缺血后,可在后续较长时间缺血中得到保护,对缺血的耐受性会明显提高,并基于此提出了缺血预处理的概念。目前,大量临床证据表明,缺血预处理对心肌损伤有保护作用[12-14]。
肥胖导致的脂肪细胞因子失去平衡是心血管疾病的重要诱因。CTRPs 是一类新发现的脂肪组织源性细胞因子,其分子结构与脂联素相似,共包含15 个成员,除CTRP4 外,其他成员均由氨基末端的信号肽、胶原样结构域、短的可变结构域及羧基末端与补体成分C1q 同源的球状结构域等4 个不同结构域构成[15-16]。李浩等[17]发现,CTRP9 在大鼠心肌缺血再灌注损伤的脂肪组织及血浆中表达水平明显下降,脂肪组织NADPH 氧化酶和血浆游离脂肪酸含量明显增加,且3T3-L1 脂肪细胞经过氧化氢或如软脂酸处理后,CTRP9 水平明显降低,表明心肌缺血性损伤可促进脂肪组织氧化应激,从而降低CTRP9 表达水平。本研究中,HE 染色结果再次验证缺血预处理可对脑损伤发挥保护作用,且CTRP9 抑制剂干扰可抑制后处理的保护效应。
缺血再灌注损伤是一种炎症反应过程,其病理生理作用包括补体激活、自由基生成、炎症因子产生、细胞凋亡等[18]。本研究通过免疫组织化学和TUNEL 检测结果证明心肌缺血再灌注可导致炎症反应级联效应和凋亡因子过表达,不仅可导致心脏衰竭,同时还会对其他脏器造成不良影响,其中脑组织受到的影响最大,严重的脑损伤会大幅度降低患者的生活质量。此外,后处理可有效减轻心肌缺血再灌注所致炎症反应和凋亡因子过表达,CTRP9可通过激活AMPK,抑制心肌细胞凋亡,发挥心肌保护作用,与NIEMANN等[19]的结论基本一致。
AMPK 是一类异源性的三聚体的蛋白激酶,具有调节物质能量代谢的作用。运动、AMP/ATP比例升高、缺血低氧及促炎因子等均可促进AMPK 磷酸化[20]。激活状态的AMKP 可通过多种途径参与能量合成、代谢,且对炎症反应具有抑制作用,可减轻机体氧化应激损伤[21]。AMPK 信号通路受损与肥胖、糖尿病及心血管疾病关系密切。心肌缺血时,心肌组织AMP 含量升高,AMP/ATP 比值升高可激活AMPK,促使AMPK 磷酸化,进而激活下游信号分子,引发相应的生物学效应。AMPK 激活可加快心肌细胞摄取、利用葡萄糖的速度,改善能量供应[22]。本研究中Western blotting 检测结果表明,AMPK 信号通路调节CTRP9 后处理的心肌保护作用,给予CTRP9 抑制剂干预可抑制该保护作用。CTRP9 抑制剂处理后,再灌注后的溶酶体功能损伤标志性蛋白P62 和自噬体标志性蛋白LC3 相对表达量升高,表明CTRP9 后处理可促进再灌注后的自噬流恢复,进而保护心肌缺血再灌注损伤,与覃琴等[22]的研究结果大致相同。
综上所述,CTRP9 对大鼠心肌缺血再灌注损伤有一定的保护作用,其可能通过激活AMPK 信号通路发挥作用,使AMPK 磷酸化来促进自噬体与溶酶体结合,加速自噬达到消化降解代谢产物再循环利用的效果,以此来发挥保护心肌缺血再灌注损伤的作用。缺血预处理时可通过调高CTRP9 水平来降低缺血再灌注损伤;应用CTRP9 抑制剂后,其保护作用将受到抑制。
