斑马鱼小头震颤突变体mise的鉴定与基因定位
2022-10-03何嘉玲王天奇暴国张长勇李楠孙德明
何嘉玲王天奇暴 国张长勇李 楠孙德明
(国家卫生健康委科学技术研究所实验动物中心,北京 100081)
原发性小头畸形(microcephaly primary hereditary,MCPH)是一种大脑发育障碍,导致头围比年龄和性别的平均值低3个以上的标准差[1]。患者可出现一定程度的智力障碍,或伴发多动症、表达性语言障碍和癫痫等症状,因此也常称为小头畸形综合征。原发性小头畸形多数基于常染色体隐性遗传模式,目前已经报道过的与小头畸形相关的基因有18个[2],是小头畸形的病因研究和诊疗的切入点。小头畸形致病机理复杂,利用单基因突变动物模型开展研究是表型发生机制良好的切入点。基因突变动物模型是基因功能生物学研究的重要工具。通过研究突变体的表现型和鉴定突变基因,能获得关于基因功能和异常发育相关的大量信息。
斑马鱼作为一种重要的模式生物,具有与人类高度保守的大脑组织区域和发育过程[3],加之早期胚胎透明易于观察,是新兴的研究中枢神经系统发育性疾病的重要工具[4]。本文作者在斑马鱼繁育过程中发现了一种可以遗传的,表现为小头、小眼、震颤的斑马鱼突变体,是典型的小头畸形综合征。在人群中,虽然遗传性小头畸形综合征相对罕见,但小头畸形致病基因编码的一系列蛋白在细胞有丝分裂的关键步骤中起作用[1],因此,建立MCPH突变的动物模型,不仅能够加深人们对该疾病本身的理解,也能提高我们对大脑发育过程的认识。目前,MCPH部分相关基因的动物模型研究十分缺乏,导致很多相关基因功能和作用机制仍无法明确。本研究通过建立家系,研究新发现的小头畸形突变体斑马鱼的遗传方式。通过基因组重测序结合BSA连锁分析以及KASP技术,研究表型相关突变基因定位,以期获得特定基因相关的小头畸形斑马鱼模型,为小头畸形发病机制研究和相关基因功能研究奠定基础。同时摸索出一套适用于斑马鱼突变基因定位的方法,为斑马鱼突变体基因定位提供可借鉴的参考。
1 材料和方法
1.1 实验动物
本研究使用的野生型TU品系斑马鱼成鱼,由北京大学生命科学学院细胞增殖与分化教育部重点实验室张博教授馈赠。India品系野生型斑马鱼成鱼由清华大学生命学院孟安明实验室馈赠。成鱼体重约350~390 mg,均为20~24周龄,雌雄各4尾。按照Monte[5]述及的方法饲养成鱼并繁育、收集受精卵。受精卵去除杂质后,放入含有1‰(w/v)亚甲基蓝的培养液中,于28.5℃培养箱中培养4 d,期间每天更换新鲜培养液。以上斑马鱼繁育及实验均在国家卫生健康委科学技术研究所实验动物中心[SYXK(京)2018-0010]完成。饲养温度为(28±1)℃,每天光照14 h,黑暗10 h。本文所述及的斑马鱼相关实验方案均通过国家卫生健康委科学技术研究所实验动物福利伦理委员会审批(NRIFH21-2104-1),所有实验均符合动物伦理学标准,严格遵循3R原则进行动物饲养和实验操作。
1.2 主要试剂与仪器
海洋动物组织基因组DNA提取试剂盒(DP324,天根生化科技有限公司,中国)。Illumina测序平台(Illumina,Inc.,San Diego,CA,USA);体视镜(Stemi 305,ZEISS,Germany);Nanodrop2000(Thermo,USA);OMEGA F Reader 96 and 384 well compatible,OMEGA F SNP分型检测仪(LGC,UK)。
1.3 实验方法
1.3.1 软件与数据库
本研究中用到的软件及数据库名称和链接如表1所示。

表1 主要实验软件与数据库链接Table 1 Main experimental softwares and database links
1.3.2 突变体生物学特性观察
突变体呈现小头,小眼表型,为了解头部和眼睛的改变情况,利用ZEN软件随机测量8尾2 dpf和3 dpfmise斑马鱼和无表型同胞的最大头宽和眼睛的面积。利用GraphaPad Prism软件对测量数据进行统计。
1.3.3 遗传分析
由于mise突变体5 d死亡,为研究其遗传方式,利用mise的亲本与野生TU进行外交产生F1代。随后待F1性成熟后进行自交,分析其子代(F2)的表型情况,计算发生表型分离的F2中有表型鱼和无表型同胞的比例。
1.3.4 突变基因初步定位
(1)DNA样品制备及重测序
为获得丰富的多态性标记,含杂合突变基因的mise亲本(P0)与野生型India品系鱼外交,获得F1。待F1培养至性成熟,通过自交获得F2。选取F2发生性状分离的两个群体:即mise突变体和无表型同胞各40尾构建极端混池。使用海洋动物组织基因组DNA提取试剂盒,分别提取mise亲本(P0),野生型India亲本,F2mise混池和F2正常表型混池的总基因组DNA,并用Nanodrop 2000定量后送百迈客有限公司用于文库构建和全基因组重测序。具体流程包括:使用S2/E210超声波发生器将提取的DNA产物进行超声波处理以产生350个碱基的片段(Covaris,Woburn,MA,USA);随后进行末端修复产生核苷酸突出物。然后,使用T4脱氧核糖核酸连接酶连接测序接头,并进行聚合酶链反应。最后将聚合酶链反应产物纯化并加载到Illumina测序平台(Illumina, Inc., San Diego, CA, USA)上进行双端测序。
为了简化描述,我们将杂合亲本P0命名为R01,将野生型India命名为R02,将F2mise混池命名为R03,将F2正常表型混池命名为R04。
(2)突变位点关联分析
主要采取对极端混池的突变位点进行批量分离分析的方法。首先为获得clean read,使用百迈客有限公司(中国北京)提供的内部Perl脚本,对原始数据进行过滤,去除低质量的reads,包括带接头reads,同时比对到多条染色体上的reads,N含量超过10%的reads,以及质量值低于10的碱基超过50%的reads。然后,使用Burrows-Wheeler Aligner[6]将这些高质量数据与斑马鱼基因组序列进行比对。使用Picard软件中的Mark Duplicate工具消除PCR重复,提 高SNP/InDel-calling的 准 确 性。利 用GATK软件检测SNP和小的InDel变异,之后过滤数据,排除其中有多个基因型、read深度小于4、混池间基因型相同、隐性混池基因并非来源于隐性亲本的4类SNP位点。对于混池间的Indel关联分析,采用同样的标准进行过滤。关联分析分别采用欧氏距离(euclidean distance, ED)算法[7]和SNPindex[7-8]方法进行。将上述两种方法取交集。最后对SNP和InDel对应的关联区域取交集作为小头小眼表型相关的候选区域。
1.3.5 突变基因精细定位
在初步定位候选区域中,筛选可信的SNP位点。根据SNP位点和侧翼序列设计PCR扩增引物,根据标记的距离合理分区段设计KASP标记。每个标记各设计F1与F2两条特异性SNP引物和一条通用引物(R)。特异性引物尾部分别添加能够与FAM荧光和VIC荧光结合的特异性序列。如1.3.4所述,分别提取、定量F2代mise个体,无表型同胞个体和mise亲本(P0),野生型India亲本全基因组为模板,进行KASP标记有效性检验,选取能有效区分野生亲本,隐性亲本和突变体的KASP标记。随后,使用能有效分型的KASP标记,对192枚F2mise胚胎进行KASP基因分型实验。按照LGC基因组公司(英国米德尔塞克斯,LGC)的方案,在384孔板中进行KASP分析。PCR反应体系为2×Taq DNA Polymerase Mix,2 μL,SNP Primer Mix(4×),1 μL;DNA样品,2 μL。PCR扩增体系:预变性,94°C,10 min,94°C,变性,20 s,61°C~55°C(drop 0.6°C per cycle),退火/延伸 45 s(10 cycle);94°C变性 20 s,55°C,退火/延伸45 s(35 cycle)。PCR扩增循环结束后,在低于40°C的环境下,利用SNP分型检测仪(LGC)读取荧光值。被动参比染料ROX(passive reference dye ROX)用于校正孔与孔之间由于反应体积误差导致的信号差异。结果数据分析软件为设备自带基因型读取软件LGC KlusterCaller software。
1.3.6 转录本序列分析
TRIzol法提取突变体mise和野生TU的总RNA,取1 μg反转录成cDNA,利用CDS全长引物PCR扩增候选基因的CDS,并送测序。最后将测序结果与参考转录本序列进行比对。引物序列见表2。

表2 候选基因CDS扩增引物序列Table 2 CDS amplification primers for candidate genes
1.3.7 Morpholino合成、注射及表型观察
委托genetool公司合成靶向terfa2号外显子的剪接抑制Morpholino及相应的错配对照,terfa-sMO:5’-GCGAACGTCACTGGAAAATATTACT-3’及terfascMO:5’-GCCAACCTGACTGCAAAATATTAGT-3’,以每个胚胎6 ng注射量分别注射单细胞期斑马鱼胚胎,并观察表型变化。
1.4 统计学方法
2 结果
2.1 突变体mise的表型分析
2 dpf时mise斑马鱼突变体出现明显的小头,小眼表型,部分胚胎体轴向背部卷曲。通过对2 dpf和3 dpf的幼鱼进行测量发现,mise的头(图1A和1E)和眼(1B和1F)的尺寸明显小于正常表型同胞(1C、1G和1D、1H)。mise平均最大头宽为(326.67±3.36)μm和(393.63±5.79)μm,正常同胞平均头宽度为(359.11±8.33)μm(n=8,P=0.0028)和(463.99±4.55)μm(n=8,P<0.0001),mise斑马鱼的头宽远小于正常同胞,差异极显著,出现了明显的发育迟滞。mise斑马鱼眼睛面积平均值分别为:(28013±1454)μm2和(23562±1670)μm2,都远小于无表型同胞(43921±1660)μm2(n=8,P<0.0001)和(55956±1258)μm2(n=8,P<0.0001),差异极显著。比较2 dpf和3 dpf,眼睛不仅没有随着发育时间的延长增大,反而出现了一定程度的萎缩(图1B、1F)。
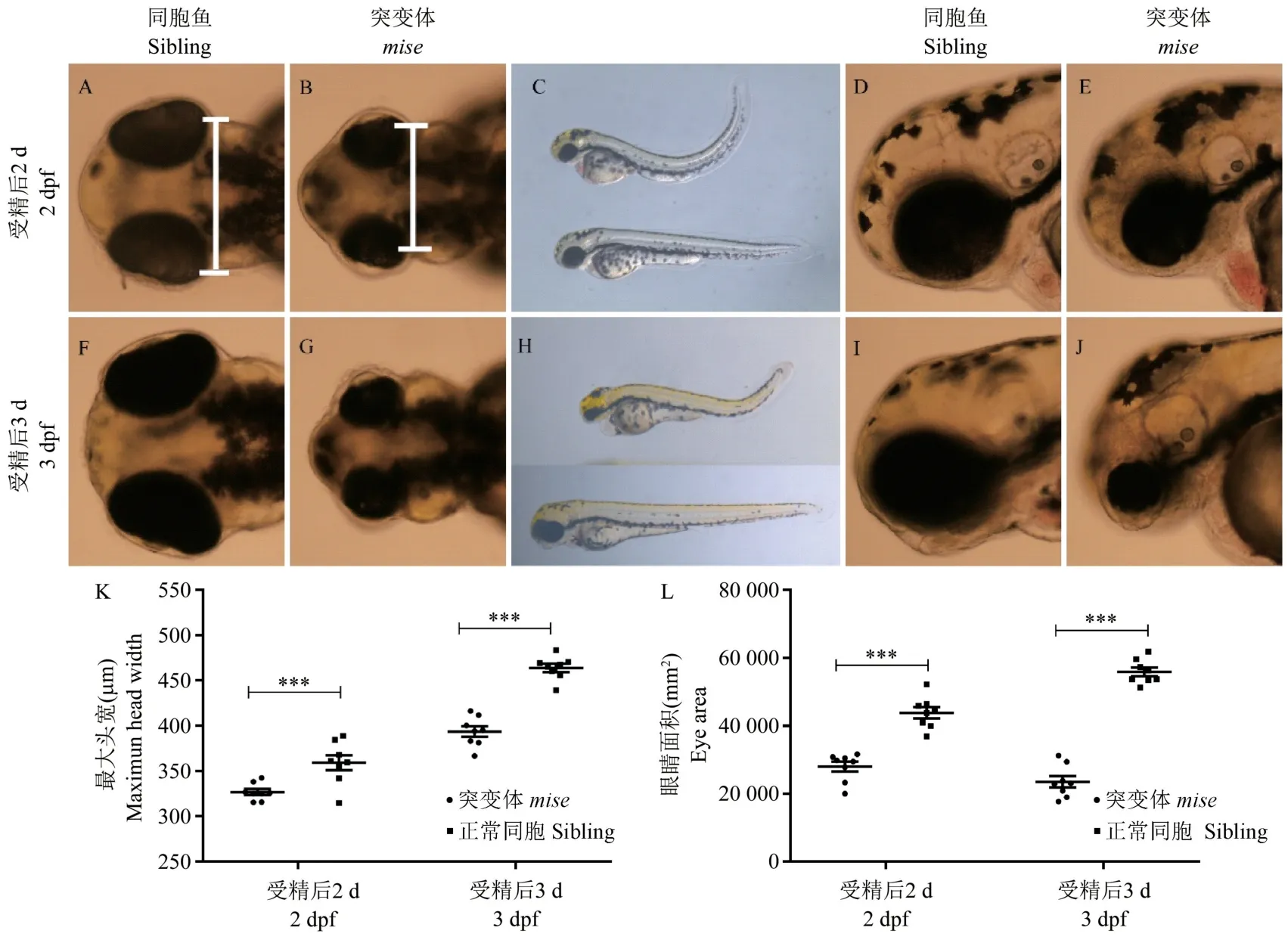
图1 mise胚胎的异常表型观察Note. A, B, D, E and F, G, I, J show the head appearance of the sibling fish and mutant with local amplification at 2 and 3 days after fertilization, respectively. C and H respectively show the overall appearance while the mutant at the top and the sibling fish at the bottom. K and L are the values of maximun head width and eye area of mutant and sibling fish at 2 and 3 days after fertilization, respectively. Compared with the control fish, ***P<0.001.Figure 1 Observation of abnormal phenotype of mise embryo
2.2 遗传规律分析
mise的亲本与野生TU进行外交产生F1代,表型分析表明,F1代所有斑马鱼表型正常。F1代斑马鱼自交,结果部分F2代表型正常,部分F2代出现表型分离。突变体2 dpf开始出现小头、小眼、震颤表型。统计3对产生F2代表型分离的F1代斑马鱼发现:第一对中,mise突变体25尾,正常表型78尾;第二对中,mise突变体35尾,正常表型105尾;第三对中,mise突变体44尾,正常表型124尾(表3)。经卡方χ2测验,该表型符合隐性基因1(隐性):3(显性)的分离比。表明F1代鱼发生了单基因隐性纯合致死突变。

表3 F2表型分离情况统计Table 3 Statistics of F2 phenotype separation
2.3 斑马鱼重测序和SNP、InDel检测
利用Illumina HiSeq平台分别对R01~R04 4个样本进行重测序分析。测序共获得211.42 Gbp数据量,过滤后得到的clean bases为209.43 Gbp。个样本测序数据Q30为89.47%~90.25%,GC含量37.72%~38.45%。样品与斑马鱼基因组(GRCz11)平均比对效率为98.27%。两亲本(F0)平均覆盖深度为17×,子代混池(F2)的平均覆盖深度为42×。总基因组覆盖度95.48%(至少覆盖1×)。过滤后得到的clean reads数分别为97471743、 106703350、238971525、 255944524(表4)。
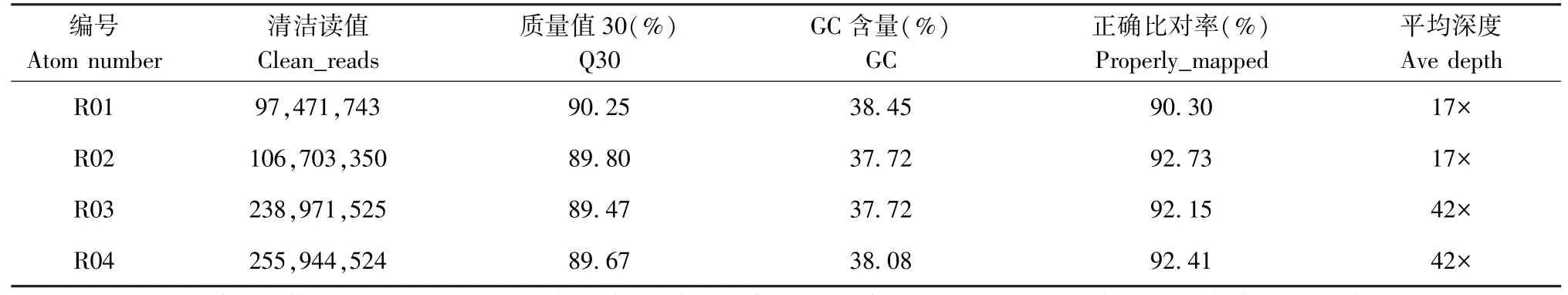
表4 测序数据质控及与参考基因组比对结果Table 4 Sequencing data quality control and results of comparison with reference genome
根据过滤条件最终筛选获得高质量SNP 3364571个,高质量的可信InDel位点778134个。具体筛选结果统计如表5、表6。

表5 SNPs过滤统计数据Table 5 Statistical datas of filter SNPs

表6 InDels过滤情况统计Table 6 Statistics of filter InDels
2.4 关联分析
基于SNP的连锁分析,按照ED方法,计算得到SNP关联阈值为0.07。关联值分布如图2A。通过对此阈值的数据分析,可在4号染色体筛选到1个性状关联区域,长度10.46 Mb,共包含564个基因,其中包含非同义突变SNP位点基因241个。
运用SNP-index关联分析方法进行计算机模拟[9]。结果表明,置信度为0.99时,在4号染色体获得1个性状关联区域,长度5.67 Mb,包含353个基因,其中非同义突变基因175个SNP-index及ΔSNP-index的分布如图2B所示。
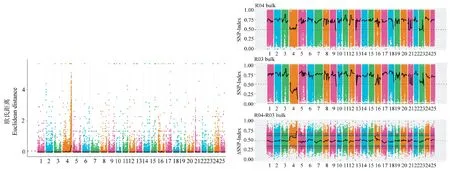
图2 SNP关联分析ED和SNP关联值在染色体上的分布情况Note. A, Abscissa is the chromosome name, the colored dots are the ED values of the SNP sites, the black line is the ED value after fitting, and the red dotted line is the significance of the association threshold. The higher the ED value, the better the association effect. B, Abscissa is the chromosome name, the colored dots are the calculated SNP-index (or ΔSNP-index) values, and the black line is the fitted SNP-index (or ΔSNP-index) value. From top to bottom are the distribution of SNP-index values of recessive pools,the distribution of SNP-index values of dominant pools, and the distribution of ΔSNP-index values. Green threshold line in the figure below has a confidence level of 0.90, and the blue threshold line the confidence level is 0.95 and the red threshold line confidence level is 0.99.Figure 2 Distribution of the associated values of ED and SNP on chromosomes in SNP correlation analysis
对于两种算法取交集,与突变关联的SNP位于Chr4:72420000~78090000,包含175个非同义突变的基因。
基于InDel的关联分析,按照ED方法,通过计算将InDel关联的显著性阈值设置为0.08。关联值分布如图3A。根据该阈值判定,在4号和5号染色体共获得2个性状关联区域,长度10.88 Mb,包含586个基因,其中包含移码突变InDel位点的基因共39个。
运用InDel-index方法进行计算机模拟[9]。结果表明,置信度为0.99时,在4号染色体获得1个性状关联区域,长度5.61 Mb,包含353个基因,其中包含非同义突变的基因共175个。关联值分布如图3B。

图3 InDel关联分析中,ED和InDel关联值在染色体上的分布情况Note. A, Abscissa is the chromosome name, the colored dots are the ED value of the InDel site, the black line is the ED value fter fitting, and the red dotted line is the significance of the association threshold. Higher the ED value, the better the association effect. B, Abscissa is the chromosome name, the colored dots are the calculated InDel-index (or ΔInDel-index) value, and the black line is the fitted InDel-index (or ΔInDel-index) value. From top to bottom are the distribution map of InDel-index value of recessive mixed pool, the distribution map of InDelindex value of dominant mixed pool, and the distribution map of ΔInDel-index value. Green threshold line in the figure below has a confidence level of 0.90 and the blue threshold line the confidence level is 0.95 and the red threshold line confidence level is 0.99.Figure 3 Distribution of the associated values of ED and InDel on chromosomes in InDel correlation analysis
对于两种算法取交集,与突变关联的InDel位于Chr4:72450000~78060000,包含30个移码突变的基因。对SNP和InDel对应的关联区域取交集,得到1个区域,总长度为5.61 Mb,位于Chr4:72450000~78060000,共包含353个基因,其中包含非同义突变的基因共174个,移码突变的基因共30个。
2.5 突变基因精细定位
由于5.61 Mb区域仍然包含大量的基因,我们开发了22个KASP标记,筛选出能有效区分亲本(P0),F2纯合突变体和无表型同胞的6个KASP标记对192个F2突变个体进行基因分型。通过KASP标记进行连锁分析,最后将与mise突变体相关的突变位点定位在Chr4∶77940617~78006239,一个65.6 kb的区域。
根据斑马鱼基因组数据库提供的基因注释信息,该区间内包含6个注释基因(表7),可能为该小头小眼震颤突变体斑马鱼的候选基因。将这些基因与BSA结果中的174个非同义突变的基因和30个移码突变基因比对,发现这里有两个LOC108183534和zgc:194336不包含在内。因此,发生非同义突变的有4个,分别为arfgap3,zgc:113921(ikbip),terfa和zgc:85975(ckap4)。

表7 定位区间内的候选基因注释Table 7 Annotation of candidate gene in the positioning interval
2.6 候选基因序列比对
为获取表型相关的候选基因,首先对候选基因的编码序列进行测序。利用扩增候选基因CDS全长的引物,扩增编码序列。通过与Ensemble中公布的参考序列比对发现,arfgap3、ikbip和ckap4发生同义突变的位置均属于SNP,因此不可能与该致死表型相关。而在比对terfa的序列时发现,mise在第二外显子发生c.236 G>A突变,密码子TGG突变为TAG终止密码子,导致翻译提前终止(图4)。
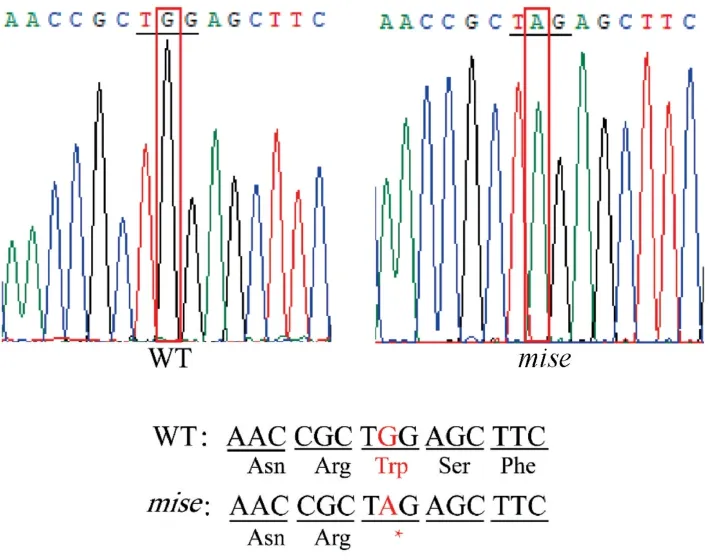
图4 mise突变体的密码子及氨基酸变化。Note. A, Sequencing peak diagrams of WT and mise mutants respectively, and red boxes indicate the mutated bases of mise and corresponding bases of WT. B, Codon and amino acid corresponding to WT and mise, and the changes of bases and amino acids are marked in red.Figure 4 Codon and amino acid changes in mise mutant
2.7 morpholino验证候选基因
基于突变体的测序结果,合成靶向terfa基因剪接抑制morpholino,注射单细胞期斑马鱼。注射3 d后观察表型发现,注射terfa-sMO的斑马鱼出现类似terfa上述点突变的小头小眼表型(图5A),成功复制了terfa缺失的表型。而注射错配对照terfa-scMO的斑马鱼,没有类似的表型改变(图5B)。说明terfa的剪接抑制的基因型变化是导致小头小眼表型的原因。

图5 WT斑马鱼注射terfa剪接抑制morpholino及对照3 d后的表型观察Note. A, Phenotype after terfa-scMO injection. B, Phenotype after terfa-sMO injection.Figure 5 Phenotype of WT zebrafish injected by terfa splicing morpholino and corresponding control 3 days after injection
3 讨论
原发性小头畸形是一种大脑发育障碍,目前已知的MCPH的致病基因参与了多种细胞过程,超过一半的MCPH的致病基因编码参与中心粒生物发生的中心体蛋白[10],部分参与DNA复制和修复[11-12],胞质分裂[13]、着丝粒功能[14]、跨膜或细胞内转运[15]以及Wnt信号自噬[16]等。可见虽然遗传性小头畸形综合征相对罕见,但对综合征的研究可以揭示对神经祖细胞、大脑大小和人脑进化的调节至关重要的分子机制。
本文作者在斑马鱼繁育过程中,发现了一个能稳定遗传的小头畸形突变体,为研究小头相关分子机制,首先要明确导致突变表型的基因。对于突变基因的初步定位,本研究采用了基因组重测序结合BSA的分析方法,快速确定了突变所在的染色体。自2013年4月英国桑格研究所(Wellcome Trust Sanger Institute)完成了斑马鱼的参考基因组的测序和全基因组分析[17]以来,已发布了11个斑马鱼基因组拼接版本,是继人类和小鼠以后,第三大高质量的有参基因组。BSA连锁分析技术,不需要构建复杂定位群体,只需要选择群体中两极端性状个体构建混池,对照参考基因组,通过计算变异(SNP,In/Del, CNV等)及其频率,即可快速定位与目的基因紧密连锁的分子标记。相较于限制性长度多态性RFLP,简单序列重复数SSR和短串联重复序列STR等分子标记方法,SNP具有数量多、覆盖密度大、遗传稳定性强、可实行批量化等优点。斑马鱼的多态性远高于大小鼠等实验动物,不同品系间的斑马鱼的SNPs高达29.9%~60%[18]。本研究通过二代测序就获得了3364571个高质量的SNP位点。为基因定位提供了丰富的分子标记。由于高通量测序的获得的变异信息量大,因此基于SNP的BSA连锁分析方法较传统的通过构建遗传图谱进行连锁分析获得的定位区间,具有关联区间范围小,定位准确,成本低,耗时短的优点。
在对目的基因进行精细定位中,本研究基于上述的高通量测序结果中SNP的二等位信息以及连锁区间,开发KASP标记对SNP进行基因分型。通过计算交换率获得与突变基因连锁的区间。KASP基因分型技术具有稳定性高、准确性高和成本低廉等优势,日益在高通量SNP分型中被广泛应用[19]。目前这种基因定位的策略主要广泛应用于农作物相关性状的基因定位[20-21]。在斑马鱼突变基因定位中也略有涉及[22],大大加速了基因定位的效率。斑马鱼突变基因的定位之所以可以套用类似的思路,与斑马鱼的基因组特点和特殊的生物学特性有关。一是,如前所述,斑马鱼存在丰富的SNP[18,23],密集的分子标记将获得更小的连锁区间;其次,斑马鱼子代数量大,在精确定位时,可以通过获得足够数量的子代分析个体的基因型计算交换率获得与性状连锁的位点。
本研究通过定位一个跟小头畸形相关的基因座,摸索了一套适用于斑马鱼表型相关基因定位的方法,为斑马鱼突变体相关基因定位提供有益的参考。也为mise基因的克隆和功能研究奠定了基础。
