Nb2NS2 材料锚固性能研究
2022-09-26零昌盛戴学琼
零昌盛,戴学琼
(桂林理工大学 理学院,广西 桂林 541004)
引言
Li-S 电池在实际应用中存在一些问题,包括循环稳定性差和库仑效率低,这主要源于硫及其放电产物Li2S2/Li2S 的低离子/电导率,以及在充/放电过程中,长链多硫化锂(Li2Sn)(n = 4,6,8)从硫电极溶解到常用电解质溶剂中(穿梭效应)。为了解决上述挑战,人们在克服硫的绝缘性能和减少Li2Sn的溶解方面做了许多努力。设计基于纳米结构的阴极,将硫限制在碳基导电框架内,包括多孔碳材料、碳纳米纤维、碳纳米管(CNT)和石墨烯[1]。Wu 等人证明了掺硼的微孔碳/硫纳米复合材料对Li-S 电池表现出超高的循环稳定性和速率能力[2]。Ai 等人制备了氮和磷共掺杂的分级多孔碳,并提出其作为硫的宿主材料的可行性[3]。此外,Jin等人表明,在CNTs 中加入B 和O 的双掺杂,可以诱发更高的放电容量,更稳定的循环稳定性,以及更好的Li-S 电池的速率能力表现[4]。MXene 是一类二维无机化合物,由几个原子厚的过渡金属碳化物、氮化物或碳氮化物层组成。各个小组已经成功地制备了一系列Mxenes,其中M 是一种早期过渡金属,A 是IIIA 组或IVA 组元素,而X 是碳和/或氮[5]。Liang 等人研究发现,Mxene 相Ti2C 作为Li-S 电池的阴极宿主材料非常有效,由于Li2Sn与表面Ti 原子的强烈相互作用,甚至在70wt%的S 下也能提供非常稳定的循环性能和高容量[6]。此外,Zhao 等人通过研究O-功能化的Ti2C和Ti3C2对多硫化锂的锚定作用[7]。结果表明,由于Ti2CO2和Ti3C2O2材料对多硫化锂的吸附能量适中,因此可以提高锂硫电池的性能。
在此,我们通过计算研究了一种通过功能化氮化铌基MXenes 捕获可溶性Li2Sn物种的锚定材料。结果表明,Nb2NS2单层可以在结合强度和Li2Sn物种的完好性之间取得平衡,可以成为具有良好循环性能的下一代Li-S 电池电极的一个有前途的候选材料。
1 计算方法和模型
密度泛函理论计算采用投影缀加平面波方法(PAW)方法进行[8],该方法在Vienna Ab-initio Simulation Package(VASP)中实现。采用广义梯度近似(GGA)中的Perdew-Burke-Ernzerho(fPBE)方法来研究相互作用电子的电子交换-相关函数[9]。DFT-D3 方案(Beck-Johnson 阻尼)被用来分析范德华(vdW)的相互作用[10]。平面波基扩展的截止能量被选择为450 eV。为了降低周期性两层材料之间的直接相互作用影响,基底的设计是采取4×4超胞和c轴方向30。的真空间距。Nb2NS2在单元格中由5 个原子组成,在4×4 超胞中由80 个原子组成。布里渊区的取样采用了由Monkhorst-Pack 方案生成的4×4×1 k 点网格,用于超胞计算,12×12×1 k 点网格用于态密度(DOS)计算[11]。所有的自洽循环都进行了迭代,直到相邻迭代步骤之间的系统总能量差小于10-5eV,并采用共轭梯度法,最大受力为0.02 eV/ A。,进行结构优化。为了获得可靠的结果,对超胞的大小、晶格常数、真空间隔、截止能量和K 点采样进行了收敛测试。
2 结果和讨论
图1 给出了Nb2NS2和LiPSs 的优化结构,如图1(b)所示,Nb2N 材料的结构是一层N 原子被两层距离为2.3。的Nb 原子夹在中间,构成三明治结构。其中每个N原子与相邻的6个Nb原子以N-Nb 键相互作用,键长为2.14。从图1(c)可以看出,Nb2N 材料在费米能级附近存在大量活跃电子,呈现出金属导电性质。为了研究Nb2N 材料对硫化锂的固定作用,以Li2S6为例计算Nb2N 材料的锚固性能,Li2S6分子遭到破坏,这是由于Nb2N 材料表面的Nb 原子对S 原子的相互作用太强,导致Li2S6分子中S-Li 化学键断裂,每个S原子直接吸附在Nb2N 表面。证明原始的Nb2N 材料不适合直接设计成锂硫电池阴极材料。为了解决Nb2NMXene 对Li2Sn相互作用过强的问题,我们对其表面进行改性修饰,引入S 原子覆盖Nb2N 表面得到Nb2NS2,S原子吸附于Nb2N表面,每个S 原子于相。邻的3个Nb原子以S-Nb键相互作用,键长为2.49A。
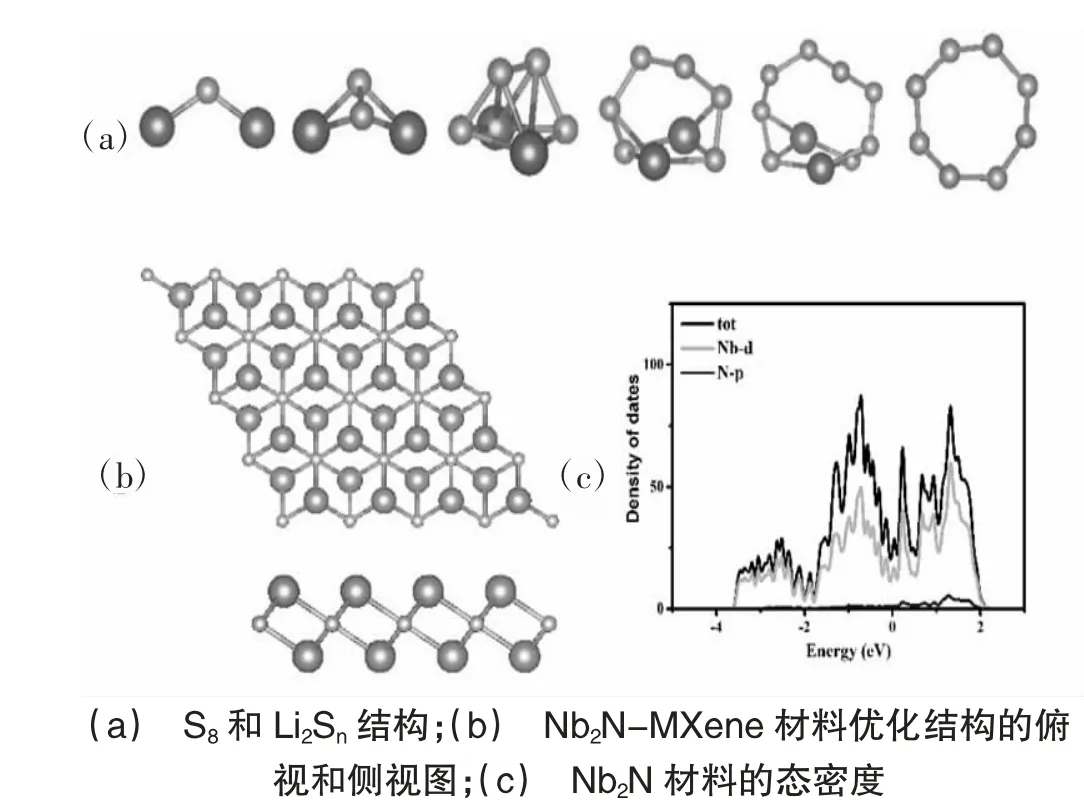
图1
我们计算了S8和Li2Sn它们之间的吸附能,并以Li2Sn分子不同朝向和不同吸附位置进行吸附,从中得到了最佳吸附结构,见图2。
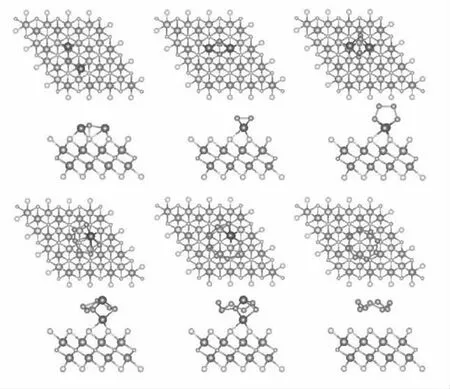
图2 S8 和Li2Sn 吸附在Nb2NS2 表面的吸附结构
从最优化结构中可以看出,S8和长链Li2Sn(n =8,6)分子更趋向于保持分子结构平行吸附在Nb2NS2表面,其中S8分子于基底材料的距离最远(达到3.51),吸附能为0.81 eV。Li2S6和Li2S8分子中的一个Li原子与基底材料表面的S 原子形成强相互作用,吸附能分别为1.36 eV 和1.26 eV。对于短链Li2Sn更喜欢分子中的两个Li 原子直接与基底材料表面的S 原子相互作用,吸附能分别为2.76 eV,1.88 eV 和1.49 eV。更重要的是,每个被吸附的硫化锂分子都结构完整。这就证明S-对Nb2N 材料表面具有改善作用,使基底材料对多硫化锂的吸附作用减弱,保证锂硫电池中充放电过程顺利进行。
为了更进一步了解Nb2NS2与S8和Li2Sn相互作用的组成成分,计算了范德华相互作用(vdW)对吸附体系吸附能的贡献,其计算结果以百分比形式体现在图3(b)中。物理相互作用的百分比表示为
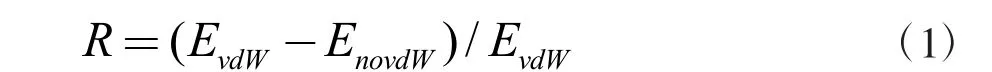
EvdW和EnovdW分别表示考虑和不考虑范德华相互作用(vdW)的系统吸附能。从图3(a)中我们可以发现,没有考虑vdW 时S8和长链Li2Sn(n = 8,6)分子吸附能很低,与考虑vdW 时的吸附能相差极大,这证明Nb2NS2材料对S8和长链Li2Sn(n = 8,6)分子的锚固作用主要由vdW 相互作用贡献,表现为物理吸附。相反的是,短链Li2Sn(n = 4,2,1)分子与Nb2NS2材料的相互作用较强,且考虑与不考虑vdW 的吸附能相差不大,这就证明vdW 修饰对它们之间的吸附能影响不大,主要由化学相互作用做贡献,表现为化学吸附。
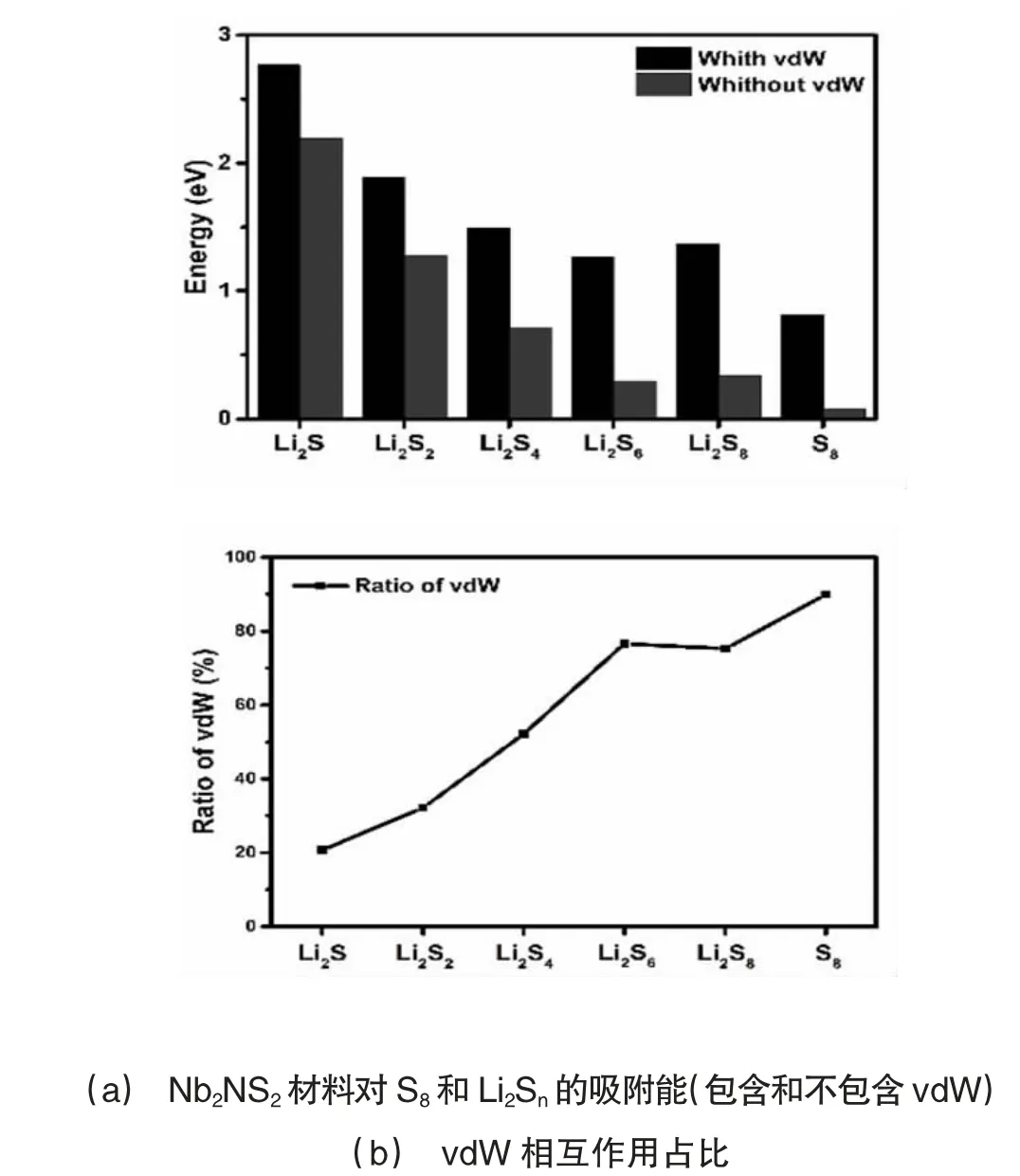
图3
为了更进一步了解S8和Li2Sn在Nb2NS2基底材料上的锚定机理,对体系进行了Bader 分析计算和微分电荷分析计算。表1 显示,在不同的锂化阶段,电荷转移量有明显的变化,S8和长链Li2Sn(n = 8,6)分子与锚固材料间的电荷转移很小,为0.08~0.12 e,短链Li2Sn(n = 4,2,1)分子与锚固材料的间的电荷转移量比较明显,为0.35~0.58 e,且总体呈现上升趋势。这证明了吸附形式从物理吸附为主转变为化学吸附为主。随着S 种和Nb2NS2材料之间的电荷转移,形成了电荷池集聚区和电荷耗竭区。因此,为了更直观的观察电荷转移情况,图4 显示了S8和Li2Sn与NbS2之间的电荷密度差,从图中可以发现S8分子与Nb2NS2之间几乎没有电荷耗尽和聚集现象,可以证明S8与Nb2NS2材料之间几乎没有电荷转移,表现为物理吸附。对于高阶多硫化锂Li2Sn(n = 8,6)体系,观察到少量的电子聚集现象,与S8分子相比,化学作用更强,但锚定主要还是来自物理相互作用的贡献,所以整体还是表现为物理吸附。对于低阶多硫化锂Li2Sn(n = 4,2,1)体系,显示出严重的电荷聚集现象,表现出强烈的化学相互作用,证明了锚固作用为化学吸附。这些结果与vdW 占比和Bader 分析的计算结果相互吻合。
表1 Nb2NS2 材料与S8 和Li2Sn 的电荷转移△Q (e),吸附能Eads(eV)和距离d ()

表1 Nb2NS2 材料与S8 和Li2Sn 的电荷转移△Q (e),吸附能Eads(eV)和距离d ()
Li�S Li�S� Li�S� Li�S� Li�S� S�ΔQ (e) 0.58 0.35 0.36 0.09 0.12 0.08 E��� (eV) 2.76 1.88 1.49 1.26 1.36 0.81 d (Å) 2.36 2.51 2.56 2.59 2.49 3.51
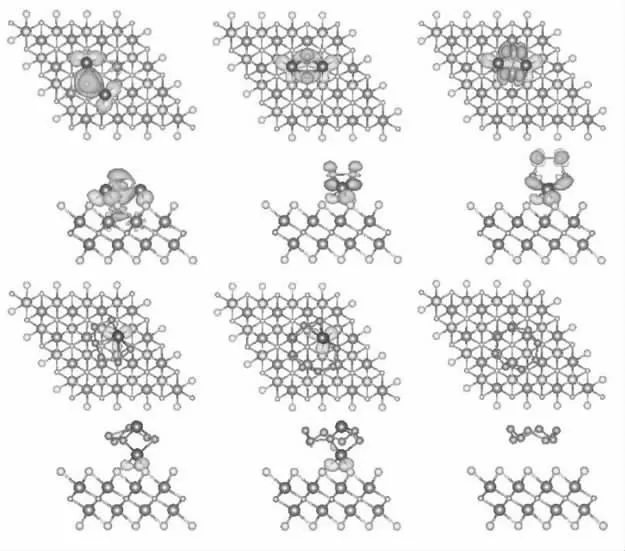
图4 S8 和Li2Sn 与Nb2NS2 之间的电荷密度差
材料导电性也是判断一种材料是否能成为锂硫电池电极材料的重要依据,因此我们计算了所有吸附体系的局部态密度。计算结果表明,对于所有体系,费米能级附近都具有大量的活性电子,很好的保持了Nb2NS2基底材料的金属导电性。良好的导电性弥补了多硫化锂导电性方面的不足,对电池的充放电速率和库伦效应有促进作用。
3 结论
通过第一性原理对Nb2N 材料锚固Li2Sn性能分析,结果表明原始的Nb2N-MXene 材料由于相互作用太强而不适合设计为锂硫电池电极材料。经过S-表面修饰,改善了Nb2N 材料表面性能,降低其对Li2Sn的吸附能至可接受范围。计算结果显示,Nb2NS2对Li2Sn的吸附能大于常用电解质的吸附能,能够有效抑制“穿梭效应”。此外,吸附前后体系的导电性能不变,呈现出金属导电性。通过我们的计算,解释了Nb2NS2成为锂硫电池电极材料的可能,为锂硫电池电极材料设计提供理论依据。
