CIC基因突变纵隔尤文样肉瘤伴多发骨转移1例报道并文献复习
2022-09-20代贺阳张丽霞徐晓飞陈凌云薛姣姣李庆霞
代贺阳,张丽霞,徐晓飞,3,陈凌云,3,薛姣姣,3,李庆霞,4△
(1.华北理工大学研究生院,河北唐山 063210;2.河北省人民医院肿瘤四科,石家庄 050051;3.河北医科大学研究生院,石家庄 050051;4.河北医科大学,石家庄 050051)
尤文样肉瘤在形态和分子上代表了一组不同类型的病变,在形态上与经典尤文肉瘤(Ewing’s sarcoma,ES)相似,免疫表型与ES有部分重叠,但缺乏ES典型的分子特征,即EWSR1-ETS融合[1]。笔者总结了1例晚期Capicua转录抑制因子(Capicua transcriptional repressor,CIC)基因突变纵隔尤文样肉瘤伴多发骨转移患者资料,现报道如下。
1 临床资料
患者,男,58岁,因“发现左侧胸壁肿物伴喘憋1周”于2020年11月25日入院。入院前1周患者偶然发现左侧胸壁一核桃大小包块,无压痛,无皮温改变,伴喘憋,活动后加重,影响睡眠,左侧卧位可缓解。既往甲状腺癌手术史4次,术后病理:甲状腺乳头状癌。查体:左侧胸壁可见约6 cm×5 cm肿物,质硬,光滑,边界欠清,活动度差。辅助检查:胸部CT(2020年11月23日)示:前中上纵隔软组织团块影(图1B),较前增大(2020年3月16日,图1A),考虑恶性占位。双肺多发转移。纵隔内多发肿大淋巴结,考虑转移。胸骨、双侧肋骨、胸腰椎椎体及附件多发类圆形稍高密度影。骨扫描(2020年11月23日,图1C):多发骨转移。胸部定位CT(2020年11月30日,图1D)示:双肺多发转移,前中上纵隔软组织团块影,较前增大。患者病情进展迅速,行1次放疗后因不能平卧停止。穿刺组织(胸壁肿物)病理(图2A、B)示:恶性肿瘤。免疫组织化学染色(图2C、D):CK Pan(+),Vimentin(+),TTF-1(—),Tg(—),Oct3/4,CD30(—),CK7(散在少量细胞+),Ki-67(活跃区40%+),LCA(—),CD117(—),S100(个别+),HMB45(—),CgA(—),Syn(—),CD56(+),CR(—),CD34(血管+),Desmin(—),Actin(—),CD99(灶状+),FLI-1(+),WT-1(+),CCNB3(—),CEA(—)。小圆细胞恶性肿瘤,结合免疫组织化学染色考虑尤文样肉瘤。多次送胸腔积液病理,查见异型细胞,未见癌细胞。为控制胸腔积液,于2020年12月9日和14日行左侧胸腔注入重组人血管内皮抑制素注射液90 mg联合顺铂40 mg治疗。结合指南推荐及患者情况,于12月10日和17日行长春新碱1 mg(第1、8天)联合多柔比星脂质体40 mg的全身化疗。胸壁肿物基因检测结果(臻和科技):BRAFp.v600E突变,CIC p.E57K突变。免疫治疗预测评估相关监测结果汇总:程序性死亡配体-1(PD-L1)蛋白表达检测(图2E):石蜡切片TPS95%CPS:95.微卫星稳定型。肿瘤突变负荷检测结果(TMB):石蜡切片6.85个突变/Mb。人类白细胞抗原(HLA)-Ⅰ类分子基因型结果:HLA-Ⅰ(A、B、C)杂合。2020年12月27日复查胸部CT提示病情进展且胸腔积液难以控制。结合基因检测报告于2021年1月2日给予卡瑞利珠单抗200 mg免疫治疗。患者后期出现低蛋白血症、电解质紊乱。2021年1月10日患者突发心搏骤停,抢救无效死亡。
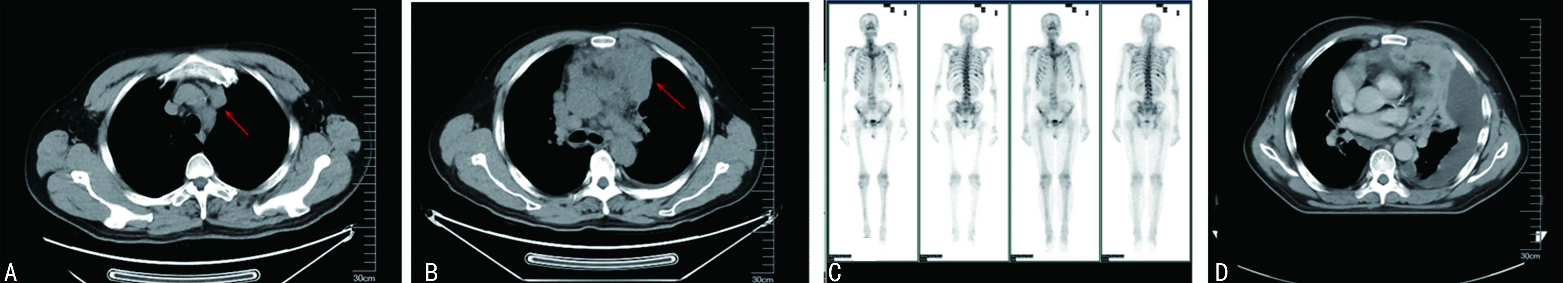
A:前中上纵隔软组织团块影(2020年3月16日);B:前中上纵隔软组织团块影增大(2020年11月23日);C:骨扫描显示多发骨转移(2020年11月23日);D:双肺多发转移,前中上纵隔软组织团块影,较前增大(2020年11月30日)。
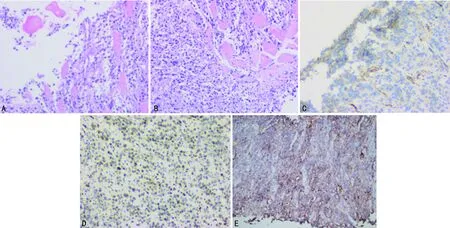
A、B:胸壁肿物活检(HE,100×);C:CD99(灶状+)免疫组织化学染色(100×);D:FLI-1(+)免疫组织化学染色(100×);E:PD-L1石蜡切片(200×)。
2 讨 论
ES是第1个被发现具有特征性染色体异位的肉瘤。分子学检测大部分存在EWSR1与ETS家族基因的融合,少部分存在FUS与ETS家族基因的融合[2]。尤文样肉瘤包括4种主要病理分型:CIC重排肉瘤(CIC-rearranged round cell sarcomas,CRS)、BCOR重排肉瘤、EWSR1与非ETS家族基因重排的肉瘤和未分化小圆细胞肉瘤。BCOR重排肉瘤比CRS预后好[3]。CRS以反复出现的CIC基因重排为特征[2]。CRS比经典ES更具侵袭性,系统治疗的效果更差[1]。BRI等[4]证实CRS通常出现在骨外部位,随着转移(主要是肺和脑)的发生,预后不良。BCOR重排肉瘤的患病率为所有未分化/未分类肉瘤的4%~14%[5]。BCOR-CCNB3融合阳性肉瘤是尤文氏肉瘤家族(Ewing’s sarcoma family of tumors,ESFT)中最常见的成员[3]。BCOR-CCNB3肉瘤更常累及骨盆、下肢和椎旁区,内脏位置极其罕见[6]。
ES诊断是基于免疫组织化学和分子遗传分析。免疫组织化学染色有助于把ES/原始神经外胚层肿瘤(primitive neuroectodermal tumor,PNET)从其他类型的小圆肿瘤细胞中鉴别出来[7]。ESFT具有特殊的染色体易位,EWS基因和编码ETS转录因子家族成员的基因融合。ES的染色体分析显示t(11;22)(q24;q12)易位在85%的病例中产生EWS-FLI1融合蛋白,在15%的病例中通过t(21;22)(q22;q12)易位形成EWS-ERG融合蛋白[8]。GUPTA等[9]认为CD99和FLI-1标记物对ES的诊断具有较高灵敏度,但缺乏特异度。CRS和ES在免疫组织化学上均为CD99阳性。CRS通常显示CD99为弥漫性染色,而不是强染色。此外,CRS通常表达ETV4和WT-1、ERG和FLI,而ES表达NKX2-2.6[10]。该患者免疫组织化学染色提示小圆细胞肉瘤,符合尤文样肉瘤且存在CIC基因突变。
ES主要发生于青少年和儿童,在所有软组织肉瘤患者中占比<1%,主要累及(约80%病例)长骨干骺端[11],骨转移可反映原发肿瘤的进展状态,出现骨转移、恶性胸腔积液常提示疾病已进入晚期。JIANG等[12]研究指出骨外ES相对于骨ES具有更强的侵袭能力,发生转移的骨外ES患者的5年生存率和整体生存率分别为24.70%和26.18%,而转移性骨ES患者的5年生存率和整体生存率分别为31.40%和32.47%。最近的研究表明,结合手术切除、大剂量化疗药物应用和高剂量放射治疗,骨外ES长期存活率已经增加至30%~40%[13-14]。局部手术联合放化疗(长春新碱、阿霉素、环磷酰胺、异环磷酰胺、依托泊苷)被认为是治疗任何部位ES的最佳方案。该患者治疗效果不佳的原因可能为:(1)患者就诊时已是肿瘤晚期,失去手术及放疗机会,且肿瘤恶性程度高,病情进展迅速,没有足够的时间接受治疗,治疗疗程不够;(2)经典的ES化疗、胸腔注药及根据基因检测报告给予免疫治疗,这种治疗方案不适合该患者;(3)电解质紊乱引起心搏骤停。
综上所述,本病例为CIC基因突变的纵隔尤文样肉瘤且PD-L1高表达。目前还没有基于分子的靶向治疗或癌症免疫治疗用于治疗CIC基因突变肉瘤的报道,细胞毒性药物的化疗仍然普遍使用[15],最佳治疗方案尚不明确。该病在成人患者中罕见,但应该被考虑为纵隔占位的鉴别诊断。了解这种罕见的骨外ES/PNET实体瘤、特征性的影像学表现和免疫组织化学特征,可以帮助临床医生对这种高度恶性的肿瘤进行正确的诊断和更好的治疗。目前,尤文样肉瘤治疗参照ESFT临床循证诊疗指南,并未对其进行更详细的分类说明,治疗效果不理想。因此,在今后临床工作中,需要为患者制订更为个体化的治疗方案,以达到延长患者的生存期的目的。
