重组Flag-pA-Tn5蛋白的原核表达及酶活性鉴定
2022-09-17田家俊夏燕李玉玲任雪谢金芳王南平黄晓飞曹春雨
田家俊,夏燕,李玉玲,任雪,谢金芳,王南平,黄晓飞,曹春雨
1.三峡大学医学院 肿瘤微环境与免疫治疗湖北省重点实验室,湖北 宜昌 443002;2.湖北民族大学附属民大医院风湿性疾病发生与干预湖北省重点实验室,湖北 恩施 445000
Tn5蛋白最初由BERG等[1]在大肠埃希菌卡那霉素抗性基因研究中发现,其编码DNA序列约5 800 bp,由核心序列和两条倒置的IS50序列(IS50R,IS50L)组成。Tn5转座酶能以其IS50序列的倒置末端结合到供体基因的转座子序列末端,并形成由一个二聚体Tn5转座酶和两分子DNA组成的联会复合体[2],由此获得切割和连接双链DNA的活性,并通过剪切和粘贴机制实现对供体基因的转座作用[3-4]。在此基础上,通过对野生型Tn5蛋白的转座酶IS50R结构域进行E54K、M56A和L372P 3个点突变,可获得具有体外转座活性的Tn5转座酶[5-6]。Tn5转座酶的剪切和粘贴机制已广泛应用于功能基因组学研究[7-10],如ATAC-seq技术使用Tn5转座酶组装形成带有测序接头的转座复合体,通过对细胞内染色质开放区域的DNA双链进行剪切和连接测序接头引物,获得染色质开放区域DNA的测序文库[11-13]。CUT&Tag是新近开发的染色质结合蛋白鉴定技术,该技术利用Tn5转座酶的转座作用和葡萄球菌蛋白A(staphylococcal protein A,SPA)对抗体的结合作用,通过制备一种PA和Tn5转座酶的融合蛋白,即可将Tn5转座酶与目的蛋白抗体结合。这种带有目的蛋白抗体的Tn5转座酶可在抗体介导下,特异性地将目的蛋白附近的开放染色质DNA片段切割并连接测序接头,从而可检测基因组范围内组蛋白修饰酶、RNA聚合酶Ⅱ和转录因子等结合的染色质区域[14-16]。
本研究旨在通过分子生物学方法获得带有Flag标签的PA-Tn5转座酶,为进一步探索Tn5转座在功能基因组学研究中的应用奠定基础。
1 材料与方法
1.1 质粒及菌株 质粒pTXB1-Tn5购自美国Addgene公司;大肠埃希菌DH5α、BL21(DE3)感受态细胞购自北京全式金生物技术有限公司。
1.2 主要试剂及仪器 几丁质树脂购自New England Biolabs(北京)有限公司;DNA限制性内切酶、T4 DNA连接酶、DNA及蛋白质分子量marker购自美国ThermoFisher Scientific公司;质粒提取试剂盒购自生工生物工程(上海)股份有限公司;酵母提取物和胰蛋白胨购自英国OXOID公司;氨苄青霉素购自北京华美生科生物技术有限公司;其他分析纯生化试剂购自国药集团化学试剂有限公司;HRP标记的山羊抗兔IgG购自武汉赛维尔生物科技有限公司;BG-gds AUTO320紫外凝胶成像仪购自北京百晶生物技术有限公司;SLPe超声波细胞破碎仪购自美国BRANSON公司;3300 mini化学发光成像仪购自上海勤翔科学仪器有限公司。
1.3 重组原核表达质粒的构建 在NCBI(https://www.ncbi.nlm.nih.gov/)网站中查找SPA的编码序列(NC_016941.1),合成双链互补的5′端带有XbaⅠ和3′端带有NdeⅠ黏性酶切末端的Flag-pA编码DNA序列(Flag编码序列:5′-GATTACAAGGATGACGACGATAAG-3′),由苏州金唯智生物科技有限公司合成。用XbaⅠ和NdeⅠ酶切消化质粒pTXB1-Tn5,琼脂糖凝胶电泳后切胶回收得到带有黏性末端的线性化pTXB1-Tn5载体;将上述插入片段和线性化载体用T4 DNA连接酶16℃连接过夜,转化大肠埃希菌DH5α感受态细胞,涂布含100 μg/mL氨苄青霉素的琼脂平板,37℃恒温培养过夜后挑取单克隆扩大培养。提取pTXB1-Flag-pA-Tn5重组质粒,进行双酶切鉴定,并送苏州金唯智生物科技有限公司进行DNA测序验证。
1.4 重组Flag-pA-Tn5蛋白的原核表达 将重组表达质粒pTXB1-Flag-pA-Tn5转化大肠埃希菌BL21(DE3)感受态细胞,涂布含100 μg/mL氨苄青霉素的琼脂平板,次日挑取单克隆,接种于3 mL含氨苄青霉素的LB液体培养基中,37℃,220 r/min振荡培养16 h;取500 μL菌液,转接至10 mL含氨苄青霉素的LB液体培养基中,扩大培养4 h至A600达0.6时,再次转接至1 L含氨苄青霉素的LB液体培养基中,37℃,220 r/min培养4 h至A600达0.6,放置降温至10℃;加入IPTG至终浓度为0.5 mmol/L,28℃,220 r/min诱导16 h;4℃,13 776×g离心15 min,收集菌体,10% SDS-PAGE结合考马斯亮蓝染色分析目的蛋白的表达。
1.5 重组Flag-pA-Tn5蛋白的分离纯化 菌体用80 mL HEGX溶液[20 mmol/L HEPES-KOH(pH 7.2),0.8 mol/L NaCl,1 mmol/L EDTA,10%甘油,0.2%Triton X-100,1 mmol/L PMSF]充分重悬后,置于冰上,用超声仪进行超声破碎(超声破碎15 s,停5 s,记为1个循环,持续20个循环);将超声裂解液于4℃,30 996×g离心30 min,吸取上清液,置于100 mL烧杯中,将烧杯置于磁力搅拌器上边搅拌边逐滴加入2.1 mL 10%聚乙烯亚胺;将上述溶液于4℃,19 837×g离心10 min,取上清液,4℃保存。取5 mL几丁质树脂填充于柱子中,加入5倍柱床体积的HEGX溶液平衡柱床,平衡结束后将预处理过的样品过柱,用200 mL HEGX溶液洗涤柱子,再用12 mL目的蛋白洗脱液[20 mmol/L HEPES-KOH(pH 7.2),0.8 mol/L NaCl,1 mmol/L EDTA,10%甘油,0.2% Triton X-100,100 mmol/L DTT]快速流过柱床,待液面与柱床相切时依次堵住下方液体流出口和上方加样口。将柱子置于4℃冰箱孵育48 h,用目的蛋白洗脱液洗脱。纯化后的蛋白用2×dialysis buffer[10 mmol/L HEPESKOH(pH 7.2),0.2 mol/L NaCl,0.2 mmol/L EDTA,20%甘油,0.2% Triton X-100,2 mmol/L DTT]进行透析复性,每12 h换1次透析液。10%SDS-PAGE结合考马斯亮蓝染色检测目的蛋白的纯化。
1.6 重组Flag-pA-Tn5蛋白的Western blot鉴定将纯化后的蛋白样品经10% SDS-PAGE分离后,采用电转法将蛋白条带转移至PVDF膜上,用含5%脱脂奶粉的TBST室温封闭1 h;TBST洗膜,加入HRP标记的羊抗兔IgG(1∶3 000稀释),室温孵育1 h;TBST漂洗后ECL法显影,化学发光成像仪拍照记录结果。
1.7 重组Flag-pA-Tn5蛋白的酶活性鉴定 按照PI CELLI等[17]的方法,取纯化后透析复性的重组FlagpA-Tn5蛋白,与含测序引物接头的转座子DNA片段组装为转座体[15]。取100 ng人外周血白细胞基因组DNA(宜昌市中心血站提供)和4 μL 5×TAPSDMF缓冲液[50 mmol/L TAPS-NaOH(pH 8.5),25 mmol/L MgCl2,50% DMF],分别加入不同稀释比例(1∶1、1∶2.5、1∶5、1∶10、1∶25、1∶50)的转座体各1 μL,用无菌去离子水补足体积至20 μL,混匀,55℃孵育7 min;加入5 μL 0.2% SDS,室温孵育7 min终止反应。取5 μL转座反应产物作为模板,以接头序列为引物(正向引物:5′-AATGATACGGCGACCACCGAGATCTACACAGCGCTAGACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′,反向引物:5′-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-3′)进 行PCR扩增,1%琼脂糖凝胶电泳检测。
2 结果
2.1 重组原核表达质粒的鉴定 重组原核表达质粒pTXB1-Flag-pA-Tn5经XbaⅠ和NdeⅠ双酶切,1%琼脂糖凝胶电泳分析可见大小约7 700和640 bp的条带,大小与预期相符,见图1。测序结果表明,重组表达质粒构建正确。

图1 重组原核表达质粒pTXB1-Flag-pA-Tn5的双酶切(XbaⅠ/NdeⅠ)鉴定Fig.1 Restriction map of recombinant prokaryotic expression plasmid pTXB1-Flag-pA-Tn5(XbaⅠ/NdeⅠ)
2.2 表达产物的鉴定10% SDS-PAGE分析显示,经终浓度0.5 mmol/L IPTG于28℃诱导16 h,可见相对分子质量约100 000的新蛋白条带,大小与带有内含肽-几丁质结合域的重组Flag-pA-Tn5蛋白一致,见图2。

图2 重组Flag-pA-Tn5蛋白表达的SDS-PAGE分析Fig.2 SDS-PAGE profile of recombinant Flag-pA-Tn5 protein
2.3 纯化产物的鉴定10% SDS-PAGE分析显示,目的蛋白洗脱液在相对分子质量约85 000处可见一条清晰的蛋白条带,大小与Flag-pA-Tn5蛋白一致,重组蛋白产量为211.1 μg/mL,纯度为84%,见图3。

图3 纯化的重组Flag-pA-Tn5蛋白的SDS-PAGE分析Fig.3 SDS-PAGE profile of purified recombinant Flag-pATn5 protein
通过检测杂蛋白洗脱液的吸光度值发现,洗脱液中蛋白含量随洗脱体积增加而逐渐减少,并最后趋于0,见图4A;当结束DTT孵育洗脱目的蛋白时,洗脱液中的目的蛋白含量呈先上升后逐渐下降的趋势,见图4B,提示本实验在纯化过程中洗脱杂蛋白和洗脱目的蛋白时均较彻底。

图4 杂蛋白和目的蛋白的洗脱峰Fig.4 Elution peaks of foreign and target proteins
2.4 重组Flag-pA-Tn5蛋白的Westernblot鉴定Western blot分析显示,在相对分子质量约85 000处可见清晰目的条带,大小与Flag-pA-Tn5预期相符,见图5。

图5 重组Flag-pA-Tn5蛋白的Western blot鉴定Fig.5 Western blotting of recombinant Flag-pA-Tn5 protein
2.5 重组Flag-pA-Tn5蛋白的酶活性 本实验获得的重组Flag-pA-Tn5蛋白与含测序引物接头的转座子DNA片段组装后,具有切割双链DNA的活性,其酶活性在一定范围内随着转座酶稀释倍数的增加而减弱,见图6。取此酶的转座反应产物,以测序接头引物进行PCR扩增,可得到250~2 000 bp的DNA片段。在稀释比例1∶1~1∶10范围内,该DNA片段大小范围随着转座酶稀释倍数的增加而增大,产物量则呈减少趋势,见图7。表明本实验制备的重组Flag-pA-Tn5蛋白具有转座酶活性。

图6 重组Flag-pA-Tn5蛋白的酶活性分析Fig.6 Analysis of enzyme activity of recombinant Flag-pATn5 protein
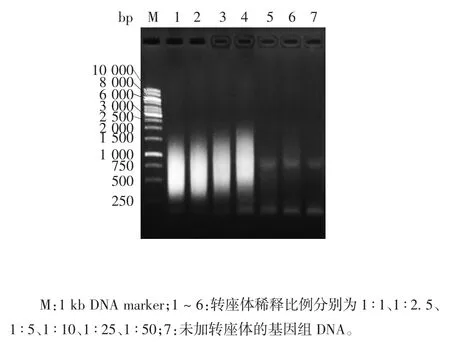
图7 转座反应产物的PCR产物电泳图Fig.7 Electrophoretic profile of transposition reaction product amplified by PCR
3 讨论
制备文库是下一代测序的关键步骤,利用Tn5转座酶对双链DNA的切割和标记机制可实现稳定且高效的测序文库构建[18-20]。使用PA标记Tn5转座酶进行CUT&Tag实验,一方面不需要甲醛交联目的蛋白和DNA复合体,有利于分离完整的细胞核;另一方面利用PA与抗体Fc段的结合,通过抗体识别、结合转录因子和组蛋白修饰酶等染色质结合蛋白,可引导Tn5转座酶对特定区域DNA序列进行标记,从而以更高的分辨率和更低的背景噪声分析特定蛋白因子对染色质结构的影响[21-22]。FACT-seq技术利用PA-Tn5转座酶对石蜡包埋组织样本进行测序,探寻人结直肠癌和人胶质母细胞瘤组织中的特异性超级增强子[23]。ACT-seq技术利用PA-Tn5转座酶在单细胞水平上绘制组蛋白修饰、组蛋白变异和染色质结合蛋白的全基因组分布[24]。WANG等[25]在CUT&Tag实验基础上进行扩展,设计出同样利用PA-Tn5转座酶的具有高通量特性和普适性的方法CoBATCH。该技术通过两次添加不同的接头引物分选捕获更多的单细胞DNA片段,利用该方法首次解析了小鼠胚胎多个器官的内皮细胞谱系发育、分化和功能的异质性。因此,获得PA-Tn5转座酶有望拓展Tn5转座酶在功能基因组学中的应用。
本实验中原核表达的Tn5转座酶C-端带有内含肽和几丁质结合结构域,实验中采用几丁质树脂柱亲和层析法对表达菌中的重组Flag-pA-Tn5蛋白进行纯化。目的蛋白上的内含肽-几丁质结合域能与几丁质树脂柱结合,而杂蛋白因无法与柱子结合而流出柱床。当还原剂DTT存在时,内含肽与目的蛋白之间的肽键断裂,经缓冲液洗脱即可得到目的蛋白。因此,纯化得到的全长蛋白相对分子质量会低于诱导表达后的内含肽融合蛋白。但纯化的重组蛋白浓度不高,后续将对试验方法进行优化,以提高重组蛋白的浓度和纯度。由于获得的重组蛋白含有PA片段,其可直接与抗体Fc段结合,因此,将IPTG诱导前后的表达菌和纯化蛋白样品经10% SDS-PAGE、转膜后直接与HPR标记的二抗孵育,对纯化产物进行鉴定。转座酶具有体外剪切和连接DNA的活性,本实验将重组Flag-pA-Tn5蛋白与人外周血白细胞基因组DNA共孵育,检测其转座酶活性。
综上所述,本实验利用分子克隆技术构建了原核表达质粒pTXB1-Flag-pA-Tn5,经IPTG诱导表达和几丁质树脂柱纯化,获得了具有良好转座酶活性的Flag-pA-Tn5转座酶,为进一步开发新的基因功能组学研究方法奠定了基础。
