以姜黄素衍生物为双配体的金属配合物的理论设计
2022-09-10郭亚星江珊林清叶孙伟明
郭亚星,江珊,林清叶,孙伟明
恶性肿瘤是严重威胁人类健康的疾病,结直肠癌(colorectal cancer,CRC)在高发恶性肿瘤中排名第三位[1]。目前已批准用于治疗CRC的靶向药物种类少且副作用大。姜黄素是一种天然多酚类物质,不仅具有抗肿瘤作用,生物相容性好,而且对人体副作用小[2]。姜黄素的作用靶点磷酸化酶b激酶(phosphorylase b kinase,PBK)[3]在CRC细胞中呈强表达[4]。姜黄素可以通过对PBK信号通路的直接抑制作用,降低PBK下游信号因子ERK1/2和H3的磷酸化表达,诱导CRC HCT116细胞凋亡[5]。然而,姜黄素自身存在抗肿瘤作用较弱、生物利用度低、水溶性差的缺点。对姜黄素进行结构修饰是提高其溶解度和生物利用度的一种有效手段。将姜黄素衍生物与金属离子结合,设计成稳定的金属配合物,有望降低金属离子的毒性和副作用,为基于天然产物的金属药物设计提供一种新策略[6]。因此,本研究拟对姜黄素进行化学修饰提高其水溶性,筛选最佳姜黄素衍生物作为配体设计新型金属配合物,考察其几何和电子结构性质,为基于姜黄素的金属配合物类PBK抑制剂的设计和合成提供理论依据。
1 材料与方法
利用量子化学计算程序包Gaussian 16[7],基于密度泛函理论(density functional theory,DFT),对姜黄素脯氨酸衍生物、双姜黄素金属配合物进行量子化学计算研究。采用GaussView[8]搭建姜黄素衍生物和配合物结构模型进行后续的结构优化。PBE0是研究过渡金属性能最好的方法之一[9],故本研究在结构优化中采用PBE0泛函。鉴于本研究涉及的分子结构较大,故在PBE0/3-21G(d)水平下对修饰后的姜黄素衍生物进行结构优化,在更高的M05-2X/6-31G(d)水平下计算其水溶性;对于姜黄素金属配合物,主族元素使用3-21G(d)基组,过渡金属Cr和Zn原子使用LANL2DZ赝势基组,在更高的PBE0/6-31+G(d)&LANL2DZ水平下计算其电子结构性质。自然键轨道(natural bond orbital,NBO)计算[10]通过Gaussian 16内置的NBO 3.1程序完成。使用 Multiwfn程序包结合VMD软件获得表面静电势(electrostatic potential,ESP)、分子轨道等图[11]。采用自洽反应场(self-consistent reaction field,SCRF)方法中的极化连续介质模型(polarized continuum model,PCM)考虑溶剂化效应的影响[12-13]。溶解能(Esol)定义为溶剂条件下优化的分子分别在溶剂条件下和气相条件下的单点能之差,即ΔEsol=ESol-EGas。采用Sybyl 2.0软件中的Surflex-Dock标准模式进行分子对接。对PBK蛋白靶标(PDB:2Y7J)进行预处理,去除对称的链和水分子,补足氢原子,再与优化后的受体药物分子进行对接打分。
2 结 果
2.1 平衡几何构型 本研究首先对姜黄素进行化学修饰,引入可改善其水溶性的基团,设计一系列新型的姜黄素衍生物。通过在水溶剂条件下对其进行结构优化,所得稳定构型如图1所示,相应的Esol列于表1。
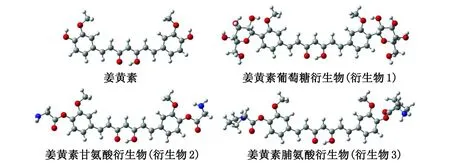
图1 在水溶剂条件下优化后的姜黄素衍生物结构Fig.1 Optimized structure of curcumin derivatives under water solvent conditions
计算结果显示,修饰后的姜黄素衍生物的Esol为-99.65~-180.87 kJ/mol,其绝对值均高于姜黄素本身的Esol(-65.73 kJ/mol)的绝对值,表明姜黄素衍生物的水溶性得到明显提高。将设计的3种新型姜黄素衍生物和姜黄素与作用靶点PBK进行分子对接,对接结果分别见表1和图2。相似性介于0~1,值越大,说明分子构象与配体结构越相似。极坐标值数值的大小反映对接结构是否合适;整体打分值最适合用于考量分子与靶标的相互作用强弱[14]。结果表明:在姜黄素中引入甘氨酸残基降低了衍生物1与靶点的相互作用,而引入葡萄糖基和脯氨酸残基显著提高了衍生物2和3与靶点的整体打分值[15](表1)。特别是引入脯氨酸的结构(衍生物3)具有最大的打分值,为9.928。打分值与配体-受体的解离平衡常数的负对数(ΔlgKd)相关,得分越高说明药物分子与受体的结合越稳定[15]。因此,本研究进一步将衍生物3中的脯氨酸置换为水溶性更好的脯氨酸衍生物,设计了8种新型化合物(图3)。

PBK:磷酸化酶b激酶。图2 姜黄素及姜黄素衍生物与作用靶点PBK(PDB代码:2Y7J)的分子对接图Fig.2 Molecular docking diagrams of curcumin and curcumin derivatives with PBK target (PDB:2Y7J)
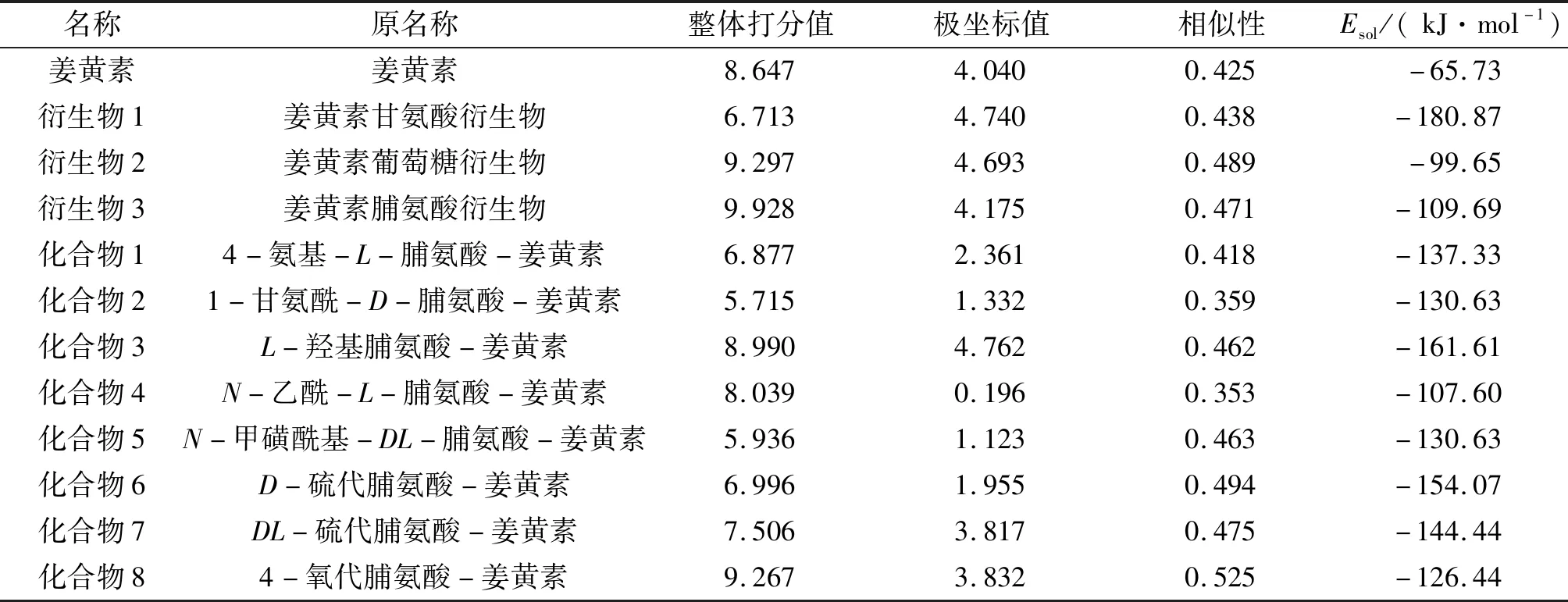
表1 姜黄素及姜黄素衍生物与作用靶点PBK分子的对接数据和溶解能Tab.1 The solvation energy of curcumin and curcumin derivatives and the docking scores of curcumin and curcumin derivatives with PBK target
所有的衍生物中姜黄素的结构基本保持了烯醇式构型(图3)。除化合物4外,其余7种化合物的Esol绝对值(126.44~161.61 kJ/mol)均大于脯氨酸衍生物3(109.69 kJ/mol)(表1)。由此可见,脯氨酸衍生物中水溶性基团的引入的确进一步增大了衍生物3的溶解度。分子对接结果表明:化合物3和化合物8对接整体打分值均优于姜黄素,说明化合物3和化合物8与PBK靶点蛋白之间的亲和力较大。因此,可以推断化合物3和化合物8可以与PBK靶点蛋白有效结合,从而降低PBK下游信号因子ERK1/2和H3的磷酸化表达,诱导CRC HCT116细胞的凋亡。值得注意的是,化合物3的Esol负值最大,为-161.61 kJ/mol,表明该衍生物溶解度最高,有望成为理想的姜黄素衍生物配体。

图3 在水溶剂条件下优化的姜黄素脯氨酸衍生物结构Fig.3 Optimized structure of curcumin proline derivatives under water solvent conditions
2.2 ZnL2和CrL2的电子结构性质 基于上述计算结果,L-羟基脯氨酸-姜黄素(化合物3)不仅具有最大的溶解度,而且具有较高的对接打分,适合作为双姜黄素金属配合物的配体。因此,本研究选择化合物3作为研究目标,解离其烯醇式结构中的一个羟基氢,得到一个-1价的去氢姜黄素衍生物阴离子,并以此为配体,与低毒性的Zn2+和Cr2+结合形成双姜黄素配体配合物ZnL2和CrL2。优化后的ZnL2和CrL2结构见图4,键长、键角等几何参数列于表2。
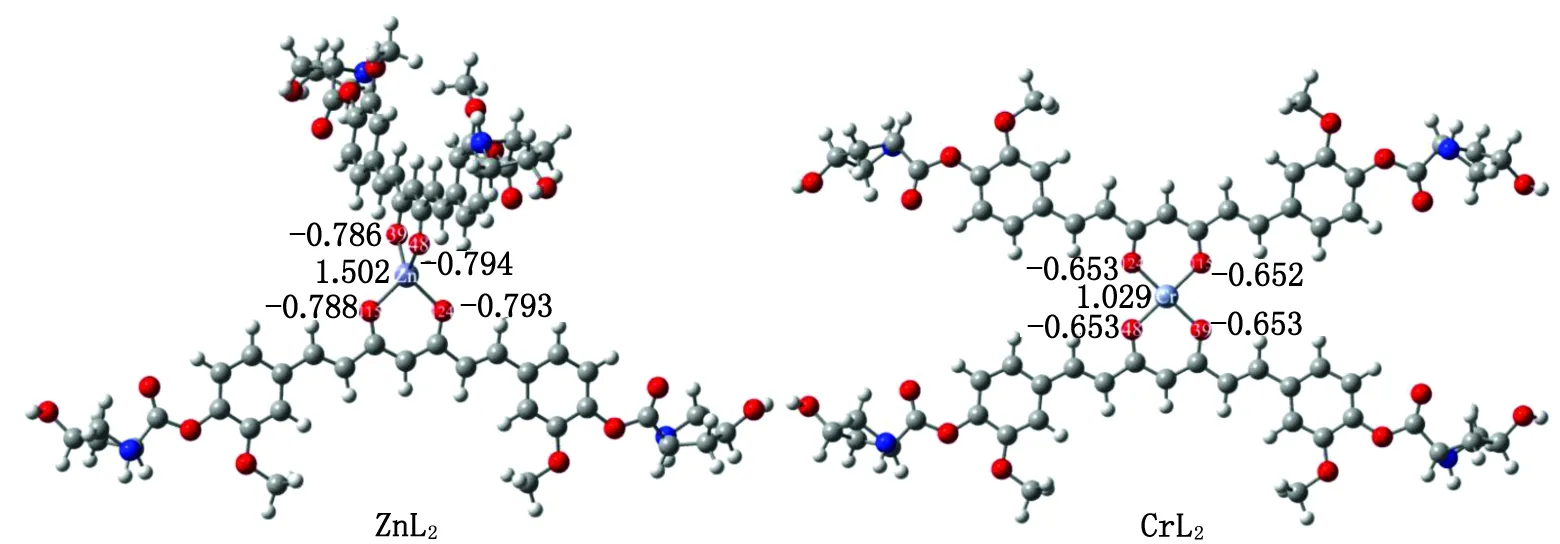
图4 水溶剂条件下ZnL2和CrL2的几何结构及其关键原子上的自然布居分析电荷(e)Fig.4 The geometry of ZnL2 and CrL2 under water solvent conditions and the natural population analysis charge (e) on their key atoms
2.2.1 几何结构和稳定性 ZnL2和CrL2中的姜黄素衍生物均保持了原来的构型。其中,ZnL2中两个配体的姜黄素分子骨架互相垂直,而CrL2中两个配体的姜黄素分子骨架与Cr原子处于同一平面。为了更详细地探究配位前后化合物3的结构变化,对姜黄素骨架的关键键长进行了对比。化合物3与Zn2+和Cr2+配位形成金属配合物后,各种键长、键角均未有较大变化(表2)。具体而言,相较于化合物3,ZnL2和CrL2中的C—O键和C—C键的键长变化小于0.002 nm,而在姜黄素与脯氨酸结合处的键角变化均<4.2°,说明化合物3引入ZnL2和CrL2之后的结构变化不大,因此其生物活性未出现较大变化。
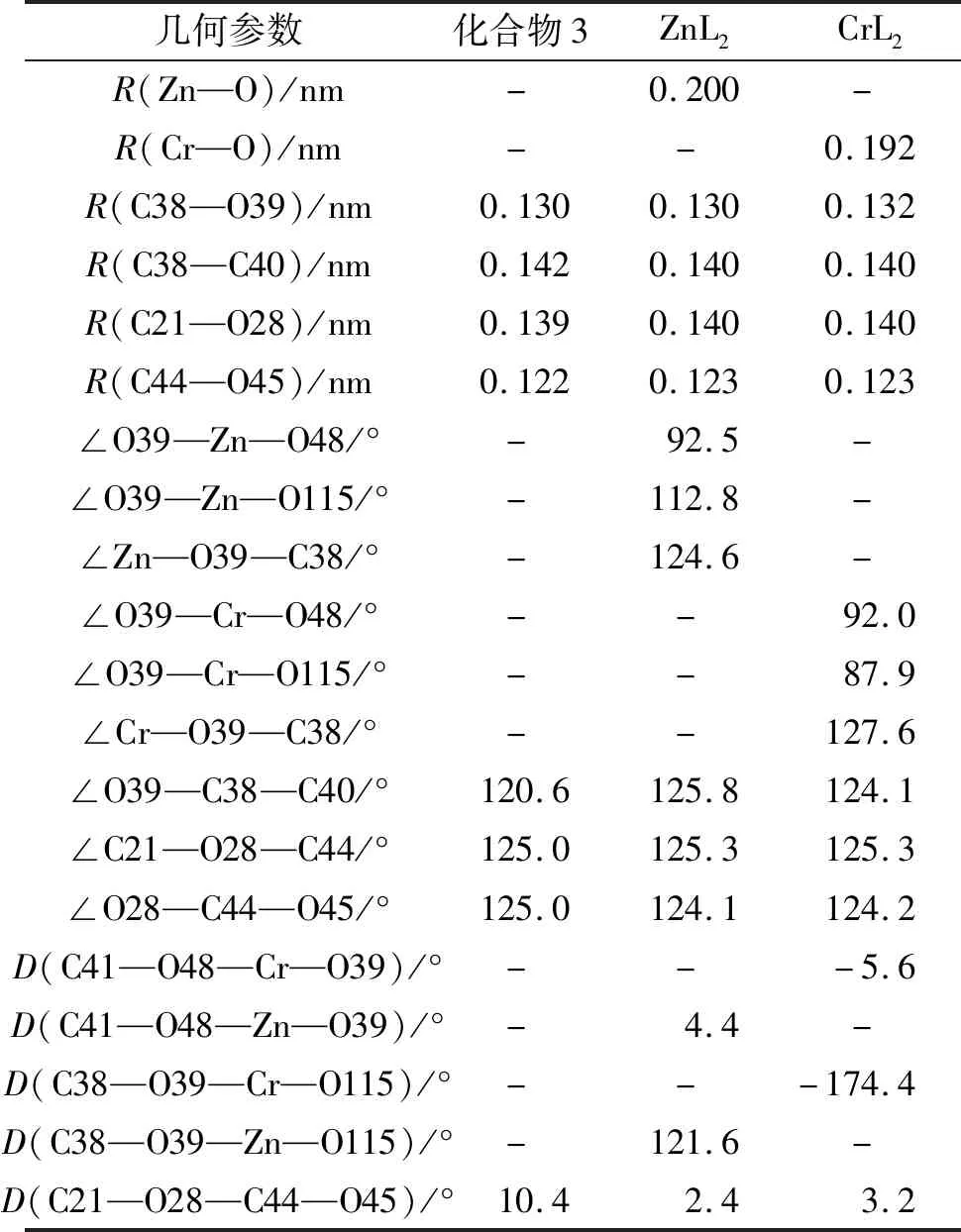
表2 化合物3、ZnL2、CrL2在气相条件下的相关键长、键角和二面角参数Tab.2 Bond lengths,bond angles and,dihedral angles parameters of complex 3,ZnL2,and CrL2 under gas phase conditions
众所周知,红外光振动谱图记录了药物的官能团在一定范围内的归属,有助于从峰的位置及形状确认配合物的形成。图5为气相中化合物3、ZnL2和CrL2的红外光谱图。由于3种药物的红外光谱的峰主要出现在500~2 000 cm-1,因此图中只给出0~2 500 cm-1范围内的光谱。从图中可以看到:化合物3在1 200~1 360 cm-1范围有多个尖锐小峰,为姜黄素衍生物的伸缩振动,而在200~1 150 cm-1有1个尖锐峰,指示姜黄素的苯环结构的伸缩振动。在1 450~1 760 cm-1范围有一些小峰,为CC键的伸缩振动。ZnL2和CrL2在1 230~1 550 cm-1范围有一些小峰,属于CC键的伸缩振动;而在1 580~1 590 cm-1有一个显著的尖峰,对应姜黄素配体的非平面摇摆振动。从该图可见,ZnL2和CrL2与化合物3的峰有所重叠,且所属峰的波长范围一致,说明化合物3在配合物中基本保持了原来的结构。由此推断,双姜黄素配体金属配合物ZnL2和CrL2的生物活性均保持姜黄素衍生物原有的生物活性。
结合能是衡量配合物稳定性的一个重要指标,可以很好地描述配体和金属核心的结合紧密程度。结合能定义为:
Eb(ML2) = 2E(L-) +E(M2+)-E(ML2)
结合能数值越大配合物越稳定。计算结果显示,ZnL2的结合能(601.85 kJ/mol)>CrL2的结合能(207.92 kJ/mol),说明ZnL2较CrL2更稳定。

化合物3:L-羟基脯氨酸-姜黄素。图5 化合物3、ZnL2和CrL2在气相中模拟的红外光谱图Fig.5 The simulated infrared spectra of complex 3,ZnL2,and CrL2 under gas phase conditions
根据前线分子轨道理论,最高占据分子轨道(highest occupied molecular orbital,HOMO)与最低未占据分子轨道(lowest unoccupied molecular orbital,LUMO)是决定体系化学稳定性的关键。HOMO轨道与LUMO轨道之间的能级差(Gap)越大,则电子从HOMO跃迁到LUMO越困难,表明体系越稳定。ZnL2的Gap值(3.67 eV)大于CrL2(2.38 eV),说明ZnL2的稳定性比CrL2高(图6)。此外,两种配合物的LUMO轨道相似,主要来自姜黄素配体的 π 轨道贡献。不同的是,对ZnL2来说,HOMO轨道的贡献来自姜黄素的 π 轨道,但CrL2的HOMO轨道贡献主要来自Cr原子的 dz2轨道。离域 π 键有助于稳定轨道,很好地解释CrL2的HOMO能级比ZnL2更高,致使其能隙值相对较小。

图6 ZnL2和CrL2的分子轨道图Fig.6 The molecular orbital diagrams of ZnL2 and CrL2
2.2.2 成键分析 Wiberg键级(wiberg bond index,WBI)分析常用于衡量共价键的强弱。本研究计算了ZnL2和CrL2中Cr2+和Zn2+与O原子形成的配位键的键级。其中,Cr2+与4个O原子形成的Cr—O键的WBI数值均为0.506,而Zn2+与4个O原子形成的Zn—O键的WBI数值分别为0.214、0.220、0.217和0.221。显然,CrL2的Cr—O键的WBI值远大于ZnL2的Zn—O键,说明Cr2+与O原子形成的共价键比Zn2+与O原子的共价键更强。因此,相比于Zn2+,Cr2+更易与姜黄素衍生物成键,即CrL2较ZnL2更加稳定。
基于分子中的原子(atoms in molcules,AIM)理论进行电子密度拓扑分析为分析化学键的本质提供了一种新的手段[16-18]。该方法是基于键临界点(bond critical point,BCP)的拓扑参数分析键的性质和强度。本研究对ZnL2和CrL2进行了AIM分析,相关参数见表3。
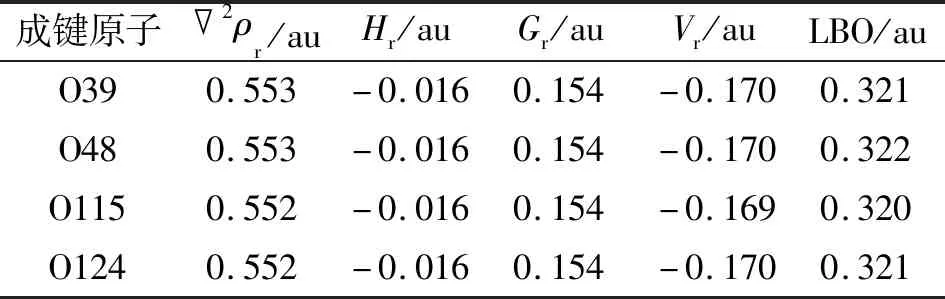
表4 CrL2中Cr—O键在BCP的拓扑参数和LBO参数Tab.4 The topological parameters and LBP parameters of Cr—O bond in CrL2 at BCP
Hr=Gr+Vr
具有共价性质的键在BCP中总是具有负的Hr,也就是说,如果2ρr>0和Hr<0,该键具有部分共价的特征[21-23]。Cr—O键和Zn—O键的Hr值为负,因此也具有部分共价键的特征。LU等[24]提出的拉普拉斯键级(Laplacian bond order,LBO)可用于描述整个键合区域的2ρr,能够很好地体现共价作用强度,避免了默认的BCP不合理导致的错误结论。这些Cr/Zn—O键的LBO值为0.118~0.322 au,清楚地证明了其共价键特征(表3~4),与上述WBI分析的结论一致。此外,Cr—O键较Zn—O键表现出更大的LBO值,表明前者比后者具有更明显的共价特征。
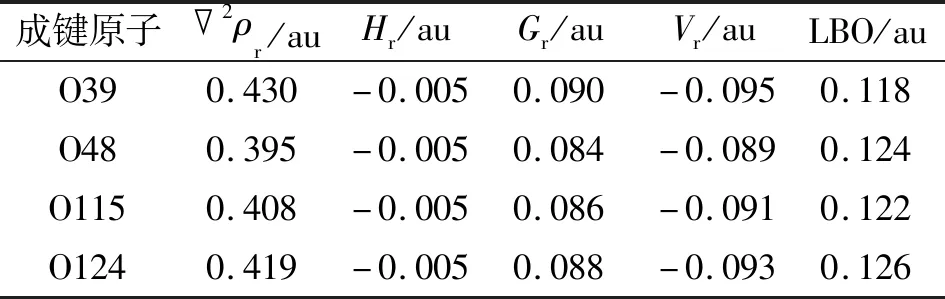
表3 ZnL2中Zn—O键在BCP的拓扑参数和LBO参数Tab.3 The topological parameters and LBO parameters of Zn—O bond in ZnL2 at BCP
为了可视化ZnL2和CrL2中Cr—O键和Zn—O键的成键本质,本研究用Boys-Foster法得到局域分子轨道(localized molecular orbitals,LMO)(图7)[25],成键原子对LMO的贡献列于一侧。ZnL2/CrL2中配位的O原子和Cr/Zn中心之间存在明显的局域电子密度,表明姜黄素衍生物与金属原子之间具有明显的共价相互作用。贡献分析表明:共享电子对主要来自姜黄素中的O原子(74.3%~77.0%),而金属原子对Cr—O/Zn—O键的共享电子密度的贡献较小(9.9%~11.4%),意味着Cr—O/Zn—O键的极性很高。对比ZnL2和CrL2,Zn原子和O原子对 Zn—O键的贡献差值(65.7%~66.2%)比Cr原子和O原子对Cr—O键(63.3%~63.4%)更大,说明Zn—O键比Cr—O键具有更明显的极性,即Zn—O键比Cr—O键具有更多的离子键成分。这也可以从NBO计算的自然布居分析(natural population analysis,NPA)的电荷得到印证[26]。如图4所示,ZnL2中O1和O2的负电荷(-0.786e和-0.794e)比CrL2中O1和O2的负电荷(-0.653e和-0.652e)更负,而Zn所带的正电荷(1.502e)比Cr(1.029e)更高,说明Zn—O键具有更明显的离子键特征。
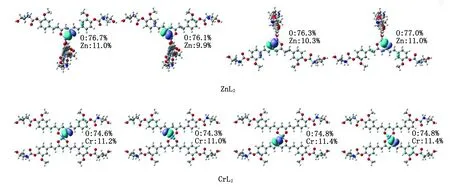
图7 ZnL2中的Zn—O键、CrL2中的Cr—O键有关的局域分子轨道Fig.7 The localized molecular orbitals of Zn—O bond in ZnL2,Cr—O bond in CrL2
分子的ESP图常用来揭示分子的电荷分布和电荷相关性质,可以指明亲电和亲核作用位点。图8 为化合物3、ZnL2、CrL2的ESP等值面图。姜黄素共轭骨架拥有离域的π电子而显示负ESP,易受亲电进攻。而姜黄素周围的氢原子和金属中心带正电,易于进攻带负电的DNA和靶标富电区域。
2.2.3 概念密度泛函理论(conceptual density functional theory,CDFT)分析 众所周知,配合物的量子化学参数对其实际应用非常重要。因此,本研究计算了姜黄素、姜黄素衍生物、ZnL2、CrL2的垂直电离能(vertical ionization energy,VIE)、垂直电子亲和势(vertical electronic affinity,VEA)和采用CDFT定义的电负性(χ)、硬度(η)、软度(S)和亲电指数(ω)等理化性质[27-29],其计算公式如下:
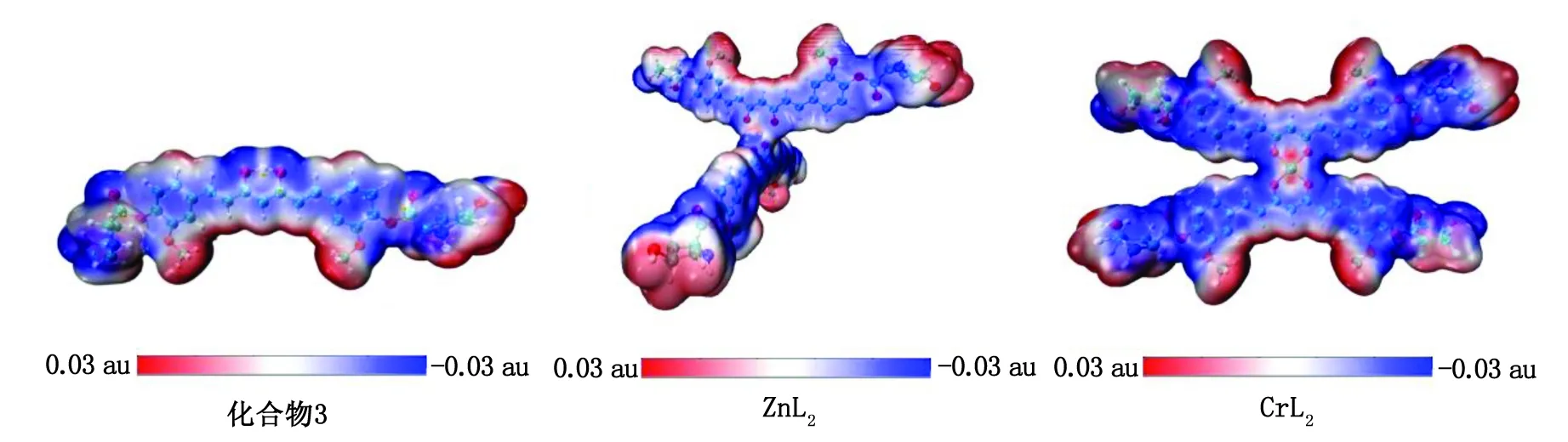
化合物3:L-羟基脯氨酸-姜黄素;ESP:表面静电势。图8 化合物3、ZnL2和CrL2的ESP图Fig.8 The ESP diagrams of complex 3,ZnL2,and CrL2
(1)
(2)
(3)
(4)
ZnL2的VIE值(5.66 eV)和η值(1.41 eV)均明显高于CrL2(表5),证明前者具有更高的稳定性。而且ZnL2的VIE、VEA和概念密度泛函参数均与化合物3和姜黄素的相关参数相近,表明它们可能具有相似的生物活性。CrL2的VEA值高达 4.89 eV,而且S和ω均为负值,说明该配合物具有更高的反应性,较不稳定。

表5 姜黄素、化合物3、ZnL2、CrL2在PBE0/6-31+G(d)水平下计算得到的相关性质参数Tab.5 The relevant property parameters of curcumin, compound 3,ZnL2 and CrL2 were calculated at PBE0/6-31+G(d) level
3 结 论
本研究基于DFT,对姜黄素结构进行化学修饰,不仅提高了分子对接分数,而且大大增强了水溶性。以筛选出来的L-羟基脯氨酸-姜黄素为配体L,设计了两种新型的双配体配合物ZnL2和CrL2,并对其几何和电子结构性质进行了系统的研究。结果表明:ZnL2具有更高的稳定性,而且保留了姜黄素及其衍生物的电子性质,最有望成为一种理想的抗CRC配合物药物分子。不可否认的是,本研究为纯理论计算工作,缺少实验佐证,因此两种配合物的生物活性亟待实验合成之后进行验证。但本研究提出了一种通过量子化学计算手段对传统药物分子进行结构改造,并将其作为配体设计新型金属药物的新思路,有望进一步推动抗癌配合物的“理性设计—实验合成—活性验证”三位一体研发,大大降低实验的盲目性。
