遗传性联合凝血因子Ⅴ和Ⅷ缺乏1例并文献复习
2022-09-06魏红媛姜春俊沈琳孔鑫垚肖志坚王玉华中国医学科学院血液病医院中国医学科学院血液学研究所实验血液学国家重点实验室国家血液系统疾病临床医学研究中心细胞生态海河实验室天津300020
魏红媛,姜春俊,沈琳,孔鑫垚,肖志坚,王玉华(中国医学科学院血液病医院&中国医学科学院血液学研究所实验血液学国家重点实验室,国家血液系统疾病临床医学研究中心,细胞生态海河实验室,天津 300020)
罕见遗传性出血性疾病(rare inherited bleeding disorders,RBD)是一组异质性疾病,约占所有遗传性凝血因子缺陷的3%~5%[1],包括遗传性纤维蛋白原缺乏症、凝血酶原缺乏症、凝血因子(F)Ⅴ缺乏症、FⅤ和FⅧ联合缺乏症、FⅦ缺乏症、FⅩ缺乏症、FⅪ缺乏症、FⅩⅢ缺乏症以及维生素K依赖性凝血因子缺乏[1-2]。2021 年9 月我院收治了1 例遗传性FⅤ和FⅧ联合缺乏的病例,现报道如下。
1 病历资料
患者,女性,46岁,主诉:间断皮肤、口腔黏膜出血30余年。患者10岁左右牙龈渗血不能自止,就诊于天津某医院,自述给予某品牌牙膏刷牙后好转。其后未诊治,皮肤黏膜及口腔黏膜间断出血,可自行停止,但出血时间较他人长。2000年顺产一女,产后大出血,给予血浆及止血治疗后好转出院。2004年顺产一子,无出血症状。2013 年因胃出血于当地医院住院治疗14 d后好转出院。2021年7月无明显诱因出现右侧臀部及右下肢大片瘀斑,就诊于兰州某医院,查凝血酶原时间(PT):20.5 s,活化部分凝血活酶时间(APTT):85.3 s,FⅤ:5.4%,FⅧ:11.5%,FⅨ:46.5%,FⅫ:
49.4 %,后患者转诊于我院进一步诊治。2021年9月3日收入我院止血血栓诊疗中心,入院查体,体温36.5 ℃,心率62次/分钟,呼吸17次/分钟。周身皮肤有出血点,无皮疹,无牙龈口腔出血,无浅表淋巴结肿大,饮食可,尿便正常。否认高血压、糖尿病、肝炎、结核等病史,无外伤及手术史,家族史不详。血常规:WBC 3.81×109/L,RBC 4.57×1012/L,Hb 134 g/L,PLT 221×109/L,肝、肾功能检测,抗磷脂抗体检测,免疫功能检测无明显异常。凝血功能检查:PT 24.8 s(参考区间10~14 s),APTT 56.0 s(参考区间22.6~32.1 s),凝血酶时间(TT)17.7 s(参考区间13.3 ~19.3 s),纤维蛋白原(Fib)2.12 g/L(参考区间2~4 g/L),抗凝血酶活性98.5%(参考区间75%~125%),FDP <2.0 μg/mL(参考区间<5.0 μg/mL),D-二聚体<0.19 mg/L FEU(参考区间0~0.5 mg/L FEU)。
2 主要检测方法及结果
2.1 凝血因子活性测定 采用一期凝固法检测原理,用CS5100全自动凝血分析仪(日本Sysmex 公司)及其配套PT、APTT试剂及乏因子血浆测定凝血因子活性;血管性血友病因子抗原(vWF:Ag)和血管性血友病因子活性(vWF:Act)测定采用乳胶颗粒增强的免疫比浊原理,用ACLTOP700全自动凝血分析仪(美国沃芬公司)及其配套试剂。检测结果:FⅤ3.7%(参考区间50%~120%),FⅧ9.1%(参考区间50%~150%),FⅡ、FⅦ、FⅨ、FⅩ、FⅪ、FⅫ及vWF:Ag和vWF:Act结果均正常。
2.2 APTT纠正实验 将患者血浆与正常混合血浆按1 ∶1比例混合,分别于即刻和37 ℃温育2 h后检测APTT,结果判断参照“罗斯纳指数”(Rosner index,RI)方法,小于10%为纠正,提示因子缺乏;大于15%为不纠正,提示存在凝血抑制物;10%~15%为临界范围[3]。APTT 纠正实验提示凝血因子缺乏,见表1。

表1 APTT纠正实验结果
2.3 Bethesda法检测FⅤ和FⅧ抑制物 将患者血浆56 ℃30 min灭活凝血因子后与等量的健康人混合血浆混合,37 ℃温育2 h检测剩余因子活性,使正常血浆因子活性减少50%定义为1 个Bethesda 单位(BU)抑制物。检测结果FⅤ和FⅧ抑制物滴度均<0.6 BU/mL(参考区间:<0.6 BU/mL)。
2.4 基因检测 留取患者静脉血液标本3 ~5 mL,采用DNA提取试剂盒(北京天根生化科技公司)提取外周有核细胞基因组DNA,Ion Torrent 测序平台(赛默飞世尔科技公司)完成高通量测序。出凝血基因突变检测结果显示:LMAN1基因检测出p.R111*纯合突变(表2)。

表2 基因突变结果
3 讨论
遗传性FⅤ和FⅧ联合缺乏症是一种罕见的凝血功能障碍性疾病,为常染色体隐性遗传,发病率约为1 ∶1 000 000,在地中海、中东和南亚国家发病率较高,可能与这些地区近亲结婚流行相关[4-5]。活化的FⅤ和FⅧ分别作为活化的FⅩ和FⅨ的非酶辅因子,参与放大凝血级联反应,FⅤ和FⅧ的缺陷导致共同凝血途径和内源凝血途径受损,实验室检查可见患者血浆PT和APTT同时延长;FⅤ和FⅧ活性水平降低,一般在正常水平5%~30%[5]。值得注意的是,虽然存在2种凝血因子联合缺乏,但是患者出血倾向并不比单一凝血因子缺乏严重,而是与同一水平单凝血因子缺乏患者相似,临床症状一般较轻[5,7],主要表现包括鼻出血、牙龈出血、皮肤淤斑、月经过多、产后出血等,而颅内出血以及消化道出血较少见。可能与FⅤ的抗凝辅因子作用相关,文献报道FⅤ通过活化蛋白C(APC)的裂解,可以形成具有抗凝辅因子功能的FⅤ,这种FⅤ与蛋白S 一起在APC 灭活FⅤa 和FⅧa中起抗凝辅因子作用,FⅤ的缺乏下调了蛋白C的抗凝作用[6]。对于患者的治疗取决于出血严重程度,主要采用输注新鲜冰冻血浆(FFP)和浓缩的FⅧ制剂,出血严重时可使用FⅦa进行止血。
FⅤ和FⅧ联合缺乏症的发病机制直到1998年才得以被揭示,是由于LMAN1(lectin mannose binding 1 或ERGIC-53)基因突变所致(本文患者即为LMAN1基因p.R111*纯合突变)。2003年,Zhang等[8]发现了第2个与该病相关的基因MCFD2(multiple coagulation factor deficiency gene 2)。文献报道LMAN1和MCFD2在内质网中以1 ∶1形成稳定的依赖Ca2+的复合物,作为特异性的载体介导FⅤ和FⅧ自内质网向高尔基体的有效转运[8](见图1)。LMAN1 和MCFD2 基因突变导致蛋白质产物的缺如或结构功能异常,从而影响FⅤ、FⅧ的正常分泌。由于该病与近亲结婚相关,所以基因检测多为纯合性突变[7]。
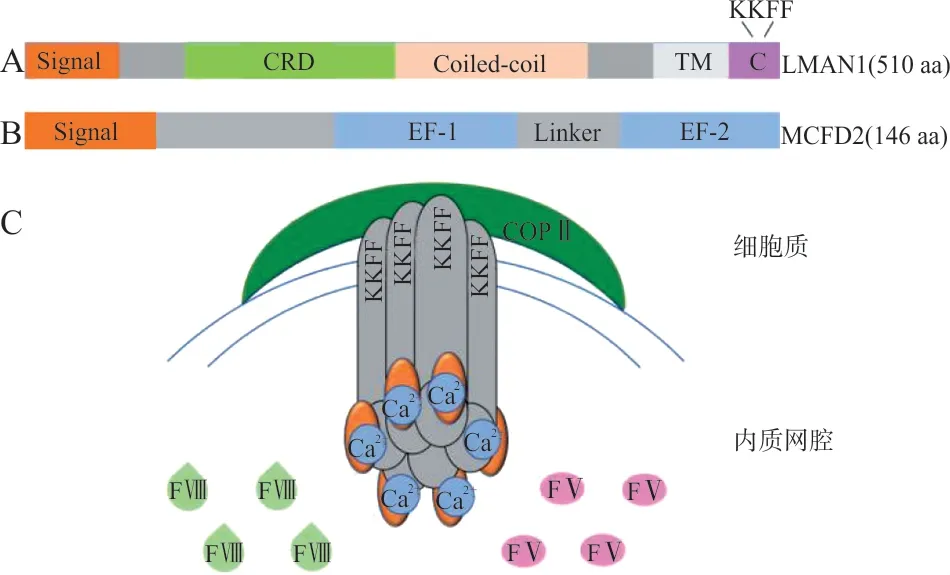
图1 LMAN1和MCFD2蛋白结构[8]
内质网中LMAN1 以六聚体形式存在,每个亚基均与MCFD2以1 ∶1形成稳定的复合物,LMAN1的CRD区与Ca2+结合后,空间构象发生变化,糖基结合位点暴露,通过结合FⅤ、FⅧ的糖基化侧链,将其富集至内质网膜区,随后与衣壳蛋白复合体-Ⅱ(COPⅡ)作用形成特异性的分泌囊泡,这一步由LMAN1的FF基序介导。囊泡离开内质网形成内质网高尔基体中间区室,FⅤ、FⅧ沿微管系统运输至高尔基体,LMAN1和MCFD2复合物则通过KK 基序返回内质网中启动下一次循环[5,7]。
本病例自幼皮肤黏膜出血,为进一步明确诊断就诊于我院,实验室检查以及基因检测结果均符合遗传性FⅤ和FⅧ联合缺乏症,诊断明确。目前由于我国基层医院缺乏相关检测手段,医务人员对本病认识不足,导致该病诊断易与轻型血友病甲混淆,或误诊为血友病甲合并FⅤ缺乏,若患者为女性时,亦可能误诊为血管性血友病伴FⅤ缺乏,但实际上该病是一种独立的疾病,与单独凝血因子缺乏的发病机制不同,希望通过本病例报道提高医务工作者对该病的认识以及为临床诊断提供参考。
