iSucc-PseAAC:基于集成机器学习的赖氨酸琥珀酰化修饰位点预测
2022-09-06贾建华吴跟强刘春生
魏 欣, 贾建华, 吴跟强, 刘春生
(1)江西服装学院商学院智慧物流教研室, 南昌 330201;2)景德镇陶瓷大学信息工程学院生物信息研究室,江西, 景德镇 333403)
蛋白质翻译后修饰(post-translational modifications,PTMs)在整个生物发展过程中,都发挥非常重要的意义[1]。它拥有着非常特殊的功能,在人类包含2~3万个基因中,只有大约2%基因编码蛋白质。其中,有一百万到两百万个蛋白质分子,是通过生物化学方式选择性剪接和翻译后修饰而成。研究发现,有许多的疾病与蛋白质翻译后修饰有关[2]。蛋白质翻译后修饰是根据转移到氨基酸残基的官能团命名的,例如:磷酸盐、碳水化合物、甲基和泛素的转移分别称为磷酸化、糖基化、甲基化、泛素化和琥珀酰化[3]。研究发现:赖氨酸琥珀酰,它在生化反应中均发挥着重要作用[4-6]。其中,琥珀酰基(-CO-CH2-CH2-CO-)和蛋白质中特定赖氨酸残基共价结合,这种变化使蛋白质的功能发生改变[7-9]。研究表明,赖氨酸琥珀酰化可能导致疾病发生,例如:肺结核。所以准确识别赖氨酸琥珀酰化位点,对研究其生物机制是非常重要的,在这一研究领域愈发受到关注。
根据目前识别赖氨酸琥珀酰化位点的回顾和比较,关于此类相关研究有许多,例如:2015年,Xu等[10]使用支持向量机建立琥珀酰化位点预测器iSuc-PseAAC;2016年,Hasan等[11]提出基于随机森林算法的预测器SuccinSite;2017年,Lopez等[12]采用了氨基酸结构特征建立SucStruct预测器;2018年,Ning等[13]结合氨基酸组成、二值编码、理化性质和灰色伪氨基酸组成等多种特征,采用基于支持向量机集成方法,称为PSuccE;2020年,Zhu等[14]使用随机森林结合了多种基于序列特征编码方法建立了Inspector预测方法;2020年,Jia等[15]使用了多种特征编码方法,并采用宽度学习预测方法建立了预测器iSuccLys-BLS,等。
这项研究中使用了多种特征提取方法,并采用了不同的集成分类器来鉴定琥珀酰化位点。并通过多次计算后,得出的数值平均作为输出结果。可以客观的评价分类器效果。
1 材料与方法
1.1 基准数据集
该研究使用了Ning等[13]构建的数据集,琥珀酰化数据来源于Hasan等[11]在UniProtKB/Swiss-Prot数据库和NCBI蛋白质序列数据库所提取的数据。使用CD-Hit[15]去除同源性超过30%的蛋白质序列,获得了2 322个琥珀酰化蛋白质。继而从2 322个蛋白质中随机分离出124个蛋白质作为独立测试集进行测试,其余蛋白质作为训练数据集。而后将实验验证的琥珀酰化位点称为阳性位点,而在同一蛋白质序列中,未被琥珀酰化位点称为阴性位点。最终采用Jia等[16]预处理数据集,具体为124个蛋白质中包含254个琥珀酰化位点和2 977个非琥珀酰化位点作为独立测试集,2 198个蛋白质中4 755个琥珀酰化位点和50 549个非琥珀酰化位点作为训练集。本研究肽链L=31,蛋白质首端(尾端)长度不够,使用X代替。原始数据集下载链接:https://github.com/weixin7112/succ。数据集见Table 1。

Table 1 Original sample data
每个样本Pδ(),可以将所有是否包含琥珀酰化位点的肽链写成:
Pδ()=R-δR-(δ-1)…R-2R-1R+1R+2…R+(δ-1)R+δ
(1)
在本研究中,总数据集分成了两部分,一类用作训练模型,一类用作测试模型。该方法又需要将数据集分为两类:
(2)
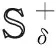
N(δ)=N+(δ)+N-(δ)
(3)
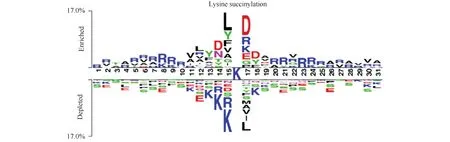
Fig.1 Two-sample Logo Statistically significant difference in location characteristics around modified and unmodified position K in the data set

Fig.2 The residue is "X" processing method The part whose color is marked in blue,the real peptide is black, and the red symbol ? in the middle indicates a mirror. indicates modification site.(A) Represents the mirror image of the δ residue X at the head of N.(B)Represents the mirror image of the δ residue X at the end of N
1.2 提取特征方法
1.2.1 One-Hot编码 当处理蛋白质序列时,通常需要将英文缩写(氨基酸)转变成为数字,这样才能形成矩阵输入到模型中训练。其中,One-Hot编码是对氨基酸序列中的每个氨基酸转变成20维度的向量,是常见的特征提取算法[19]。第一步将20种氨基酸进行编码,丙氨酸(Alanine)的编码为10…0(20维)、半胱氨酸(Cysteine)的编码为010…0(20维),…,酪氨酸(Tyrosine)的编码为0…001(20维)。本研究中,一条蛋白质序列长度L=31,则该条蛋白质序列可形成一个31*20维度的特征矩阵。
1.2.2 氨基酸组成成分(AAC) 每条序列样本中氨基酸之间存在相互关系,Nakashima等[20]研究发现,通过计算每条序列样本中20种常见的氨基酸和未知氨基酸X出现的频率,生成21维度的特征矩阵表述每条样本信息。假定一条蛋白质序列为R,长度为L,f(Ri)是序列R中氨基酸出现的次数,则每个氨基酸特征可表示为:
(4)
其中P(Ri)代表的是每条氨基酸出现的频率,i代表的氨基酸(A、C、…、Y、X),最后,可将该条蛋白质序列R用特征表示为:
以SPSS19.0软件验证涉及的临床数据,以率(%)的形式阐述计数资料,予以χ2检验,以(均数±标准差)形式阐述计量资料,予以t检验,P<0.05,统计学展现对比差异。
P(R)=[P(R1)P(R2)…P(R21)]
(5)
1.2.3 耦合序列(PseAAC)特征提取 根据Jia[18]提出的PseAAC方法,每条蛋白质序列可通过该方法表示成特征序列,首先将公式(1)表示为:

(6)
其中,Pδ()可具体表示为

(7)
和
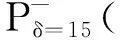
(8)
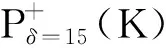
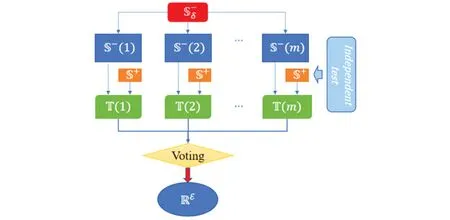
Fig.3 Classifier flowchart Flow chart of ensemble classifier voting
1.3 集成学习算法
集成学习算法已经广泛的运用在机器学习领域。通过使用多个分类器解决非平衡数据的相关问题,同时还能显著提升模型的泛化能力。在此次研究中,训练集正负样本数量分别为4 755和50 549,因此,本文采用集成学习来解决此次研究中数据不平衡问题。
支持向量机(support vector machine,SVM)和随机森林(random forest,RF)在生物信息领域中,已被广泛应用[18,21,22]。这类算法也可以运用在不平衡的数据集。本研究使用Stacking集成学习算法,建立整体的分类器,能够显著提升分类器预测准确率[23],公式如下:
(9)
1.4 评价指标
使用传统分类模型,结果一般包括了4个指标:特异性(Sp)、敏感性(Sn)、准确性(Acc)和马修斯相关系数(MCC)[24],以评价分类模型的效果,具体公式为:
(10)
接收器工作特性曲线(receiver operating characteristic curve,ROC),是反应敏感性与特异性之间的关系曲线[25]。绘制完曲线后,会对分类模型有个定性量化分析,定义为ROC曲线下与坐标轴围成的面积(Area Under Curve,AUC),AUC面积越大,说明该分类模型效果越好。
2 结果
2.1 采用不同方法对独立测试集进行5次交叉验证,得到性能指标
本文中使用了3种特征提取算法:One-Hot编码、氨基酸组成成分(AAC)和耦合序列(PseAAC)特征提取,并且使用了集成支持向量机和集成随机森林对训练集进行五折叠交叉验证。Table 2对训练集在不同方法下进行的5次交叉验证,得到性能指标。

Table 2 The training set is cross verified five times under different methods to obtain the performance index
独立测试集测试对分类模型进行评估,结果正如Table 3所示。结果发现,基于耦合序列(PseAAC)特征提取方法,集成支持向量机模型4个评价指标都是最优的,在此将该方法命名为iSucc-PseAAC。同时为了验证iSucc-PseAAC模型的稳定性,通过独立测试集评价的性能指标,可在ROC曲线(Fig.4)更直观的表示:

Table 3 The independent test set is cross verified five times under different methods to obtain the performance index
2.2 与现有方法进行比较,后续对赖氨酸琥珀酰化修饰位点预测提供较大帮助
本文将iSucc-PseAAC预测方法下的独立测试集与其他预测器做了比较,各项指标的最大值使用字体加粗表示。进一步说明本研究的有效性及实际意义,结果如Table 4所示。

Table 4 iSucc-PseAAC performance comparison with the previously proposed predictor
在这项研究中,本文通过iSucc-PseAAC整体的集成分类器,严格鉴定序列是否被琥珀酰化。iSucc-PseAAC分类器的结果表明,此次研究是有意义的,预测结果分别是:Sn = 0.827、Sp = 0.753、MCC = 0.343、Acc = 0.759。
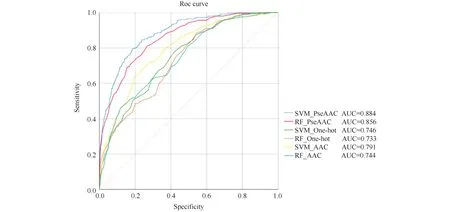
Fig.4 ROC curve of the independent test set ROC curve of independent test set under different methods
本文研究的数据样本正负比例约为1∶11,数据极度不平衡。在iSucc-PseAAC方法下,对比PSuccE和GPSuc的Sp和Acc的准确率虽然略有下降,但是在研究此类非平衡数据相关问题中,Sn和MCC是主要的考量指标,这2个指标的提升尤为重要。这两项数据对比,目前效果最好的是iSuccLys-BLS和GPSuc预测器,准确率分别提升了0.104和0.047,效果明显。与此同时,iSucc-PseAAC分类器中的Sp和Acc效果也比较好。在后续对赖氨酸琥珀酰化修饰位点的预测上提供了较大的帮助。
综合来说,iSucc-PseAAC方法对比现有方法有了稳定的提升,同时将该方法源代码已上传至Github,以供各位专家学者们参考,下载链接:https://github.com/weixin7112/iSucc-PseAAC。
3 讨论
本文的研究基于支持向量机和随机森林分类算法,使用了多种特征提取方法进行测试的同时,集成学习算法也解决了正负样本不平衡的问题,最终开发了一种iSucc-PseAAC分类器,在与其他现有预测器对比显示,iSucc-PseAAC分类器在鉴定赖氨酸琥珀酰化位点具有更优的性能。最终独立测试集预测精度分别为:Sn=0.827、Sp=0.753、MCC=0.343、Acc=0.759。
本文利用蛋白质序列特征的相关性,首先使用One-Hot编码和氨基酸组成成分(AAC),分别在蛋白质单条序列中提取特征信息,这样得到的信息是有限的,导致预测的效果并不是理想。因为样本与样本之间,会存在某种相关联系,外加此次研究数据中,样本数据量大,需要充分挖掘序列与序列信息,可能会有不错的效果。因此,本文使用了耦合序列(PseAAC)特征提取算法,充分利用了各条蛋白质序列之间存在的相关性,通过实验证实,预测的效果是显著的,对比现有的分类器,提升效果明显。
在后续的工作中,可以结合氨基酸理化性质和多种分类算法一起研究,充分挖掘样本中隐藏信息,同时也要应对少数样本中信息较少的情况下,如何鉴定修饰位点也是一项挑战。不过在生物信息统计研究中,非平衡数据研究一直是当下研究的热点,集成学习算法可以有效的提升分类预测的精度,本次研究可供各位学者提供参考。
