钠离子通道NaV1.7与神经病理性疼痛
2022-09-06王冬梅
沈 婷, 王冬梅
(福建师范大学生命科学学院,福建省发育与神经生物学重点实验室, 福州 350117)
神经病理性疼痛(neuropathic pain, NPP)属于慢性继发性疼痛[1]。NPP由多种神经类型损伤、基因突变、疾病(糖尿病、癌症等)或一些药物的毒副作用引起,临床常表现为痛觉过敏(hyperalgesia)、痛觉超敏(allodynia)和自发性疼痛(spontaneous pain),持续时间长,危害大。NaV1.7是钠离子通道(NaV channels,NaVs)α亚基亚型之一,具有通道激活电压依赖性超极化偏移[2]、慢关闭态失活和缓慢从失活中恢复[2, 3]的特点。它能够产生大量的斜坡电流[4],降低动作电位产生的阈值,放大外来小的、缓慢的去极化电流(例如感觉发生器所产生的电流),提高神经元对外来刺激的敏感性[5]。NaV1.7通道在痛觉感受器中特异性表达,是痛觉主要伤害性感受器的特异性电压门控钠通道[6]。随着研究的不断深入,NaV1.7在人类遗传学中的作用被逐渐发现,NaV1.7通道的功能获得性突变和功能缺失性突变与遗传性或自发性疼痛综合征有关,为治疗疼痛创造新型镇痛疗法开辟了新的研究方向[7-10]。目前,已有的NaV1.7选择性阻断剂具备有效的镇痛作用,且无明显副作用或成瘾问题[11]。然而,由于NaV通道亚型的氨基酸序列之间存在高度同源性,寻找NaV1.7选择性配体极为困难[12, 13];部分NaV1.7选择性抑制剂在不同类型的疼痛中需要充分暴露结合位点时才能发挥抑制作用,具有极强的状态依赖性[14];NaV1.7在不同因素引起的神经病理性疼痛的作用机制研究仍然有限。本文就NaV1.7与不同因素引起的神经病理性疼痛关系作一综述。
1 NaV1.7钠通道的分子结构及其分布
电压门控钠离子通道(voltage-gated sodium channels,VGSCs)是一种跨膜蛋白质,由1个α亚基和1或2个β亚基组成,主要负责产生Na+电流。Na+电流是动作电位的起始和传播基础,在细胞的电信号转导中具有重要作用。目前,钠离子通道已鉴定出10种α亚基亚型(NaV1.1~9和NaVX)。其中,NaV1.1、NaV1.2、NaV1.3、NaV1.4、NaV1.6和NaV1.7通道能被低浓度河豚毒素(tetrodotoxin,TTX)阻断,属于河豚毒素敏感型(TTX-sensitive,TTX-S),而NaV1.5、NaV1.8和NaV1.9属于河豚毒素不敏感型(TTX-resistant,TTX-R)。
NaV1.7钠通道是由1个α亚基和2个β亚基(β1和β2亚基)组成[15]。其中,α亚基(由SCN9A基因编辑)是由4个大型单链多肽形成的4个同源结构区域(DⅠ、DⅡ、DⅢ和DⅣ),通过细胞内连接器连接而成。每个同源结构区域内包含6个跨膜螺旋片段(S1~S6)。S1~S4段构成电压感应结构域(voltage-sensing domain,VSD),其灵活性主要由S4段螺旋结构中1/3残基上带正电荷的精氨酸和赖氨酸残基的移动介导。S5、S6段和胞外连接孔环(p -loops,p-环)形成孔隙域(pore domain,PD)[12]。孔隙域包括通道孔和选择性过滤器(selectivity filter,SF),通道孔中存在多个离子结合位点,SF由DⅠ中天冬氨酸(D)、DⅡ中谷氨酸(E)、DⅢ中赖氨酸(K)和DⅣ中丙氨酸(A)(DEKA)组成,选择性通过水合钠离子[12, 16]。NaV1.7钠通道的β1和β2亚基分别由SCN1B和SCN2B基因编辑[12],在调节通道动力学和细胞表面通道表达方面发挥着关键作用,是α亚基的辅助亚基[12]。α亚基中VSD检测到的细胞膜电位决定NaVs的开放状态。当细胞膜电位处于超极化静息电位时,NaVs大部分处于未开放状态;当细胞膜电位处于去极化状态时,电压感应结构域的运动引起α亚基构象变化,从而导致Na+选择性通道孔的开放[17];随后几毫秒内,开放的NaVs过渡到1个不导电的灭活状态。相比之下,NaV1.7钠通道中,当细胞膜电位去极化数秒后,就会出现一个缓慢的通道失活,NaV1.7钠通道β亚基辅助α亚基开放PD,从而调节电流的再生,表明NaV1.7钠通道可以参与神经元的再生电流调节[18]。
NaV1.7钠通道优先特异性表达于背根神经节(dorsal root ganglion, DRG)神经元小直径c纤维[4]上,在三叉神经节[19]、交感神经元[3]、嗅觉感觉神经元[20]以及脊髓浅层传入神经元[12]中均有分布,与其在疼痛中的作用一致。然而,与灵长类动物中NaV1.7分布不同,啮齿动物的垂体、肾上腺以及离散的脑区(例如下丘脑/视前区、穹窿下器官和几个脑干核团)中检测出较强的NaV1.7表达水平,NaV1.7基因整体缺失小鼠出生不久后死亡[21, 22],暗示NaV1.7分布存在种属特异性。
2 NaV1.7与神经病理性疼痛
2.1 NaV1.7在神经直接损伤诱发神经病理性疼痛中的作用
坐骨神经分支选择性损伤(spared nerve injury, SNI)模型和慢性坐骨神经压迫损伤(chronic constriction injury of the sciatic nerve, CCI)模型是广泛应用的2种动物神经疼痛模型,SNI模型大鼠[23]和CCI模型大鼠[24]L4和L5脊神经节(DRG)中NaV1.7表达水平升高,并伴有痛觉过敏的产生。若微量注射miR-182激动剂或过表达miR-30b,可有效抑制SNI引起的NaV1.7表达上调,缓解机械性疼痛[23]。使用白藜芦醇[25](3,4′, 5-trihydroxystilbene,3,4′,5-三羟二苯乙烯)激活沉默调节因子1[26, 27](silent information regulator 1, Sirt1),上调DRG神经元中miR-182表达,也能抑制CCI模型大鼠DRG神经元中NaV1.7表达,缓解神经病理性疼痛[24]。且miR-182[23]、miR-30b[28]属于microRNA(miRNA),在体外研究中,miRNA被证实与NaV1.7通道编码基因SCN9A基因直接配对,可作为基因表达的重要调控因子,参与调节NaV1.7转录后修饰表达。暗示miRNA通过调节NaV1.7表达缓解痛觉过敏可在多种神经病理性疼痛中实现。
临床上,降钙素能够缓解多种神经病理性疼痛,例如椎管狭窄、糖尿病神经病变和复杂的局部疼痛综合征等。CCI模型大鼠DRG神经元中,NaV1.7通道表达恢复正常可能是降钙素缓解痛觉过敏的机制之一。但有趣地是,CCI模型大鼠DRG神经元中降钙素受体未被激活,提示降钙素引起CCI模型大鼠DRG神经元中NaV1.7表达恢复正常,可能是降钙素产生某种未知信号作用于周围神经组织中引起的[29]。啮齿动物椎间盘穿刺模型大鼠椎间盘DRG神经元中NaV1.7通道[30]以及降钙素基因相关肽[31](calcitonin gene-related peptide,CGRP)等神经肽被上调,使用抗NaV1.7抗体可降低椎间盘DRG神经元中CGRP表达,从而减缓疼痛,表明椎间盘DRG神经元中NaV1.7可抑制CGRP表达达到镇痛作用,抗NaV1.7抗体是控制腰椎间盘退变患者疼痛的潜在治疗靶点[32]。以上结果表明,NaV1.7与降钙素之间的联系,将有助于深入研究NaV1.7在多种神经病理性疼痛中的作用机制。
脊神经结扎(spinal nerve ligation, SNL)模型大鼠损伤脊神经(L5DRG神经元)[33]中和眶下神经慢性缩窄环术(chronic construction injury of the infraorbital nerve,ION-CCI)模型大鼠[19]三叉神经(trigeminal neuralgia, TN)中NaV1.7表达水平下调。但脊神经结扎模型大鼠中,相邻未损伤DRG神经元(L4DRG神经元)中NaV1.7显著上调,损伤的DRG神经元(L5DRG神经元)中仍能明显观察到NaV1.7的转录物,且多聚集在L5脊神经受损端[34]。由于SNI模型中单侧神经损伤后,同侧DRG神经元中NaV1.7表达增加,能够激活对侧DRG神经元中卫星胶质细胞,导致对侧DRG神经元中NaV1.7表达增加,引起对侧DRG神经元的兴奋性增加,从而产生了镜像疼痛,表明NaV1.7可能参与了原发性传入神经自发放电的产生和传播[35]。
除此之外,SNI模型部分DRG神经元亚群中坍塌反应介导蛋白2(collapse response mediator protein 2, CRMP2)磷酸化可促进NaV1.7表达上调,维持神经病理性疼痛的产生[35]。有研究指出,磷酸化后的CRMP2可能是通过CRMP2/ Numb(endocytic proteins Numb)/表皮生长因子受体底物15(epidermal growth factor receptor pathway substrate 15, Eps15)/神经前体细胞表达发育下调蛋白4-2(neural precursor cell expressed developmentally down-regulated 4 ligase, Nedd4-2)通路调控Nav1.7的表达[36, 37]。若单独上调SNI模型大鼠DRG神经元中泛素E3连接酶(Nedd4-2)表达,能够显著降低DRG神经元中NaV1.7通道的表达[38],但Nedd4-2蛋白[38, 39]、Numb和Eps15蛋白均难以实现对NaV1.7表达特异性的调控[40, 41]。当周期蛋白依赖性激酶5(cyclin-dependent kinase 5, Cdk5)在S522位点磷酸化CRMP2时,抑制CRMP2泛素化[42]或干扰CRMP2与小泛素样修饰能够有效控制NaV1.7的转录和活性变构,达到对神经病理性疼痛的保护作用[43]。此外,使用邻(羧甲基)羟胺半盐酸盐((aminooxy)acetic acid hemi hydrochloride,AOAA)抑制胱硫醚-β-合成酶(cystathionine-β-synthetase,CBS)介导的内源性硫化氢(hydrogen sulfide,H2S)生成,通过MEK/ERK信号通路可显著降低SNI模型大鼠DRG神经元中NaV1.7通道的表达,降低Na+电流密度,减少NaV1.7电流振幅,从而达到镇痛作用[44]。
总之,NaV1.7与痛觉感受器异常兴奋性密切相关。神经损伤引起它们的分布、表达、和/或生物物理性质的改变,从而有助于神经病理性疼痛的产生和维持[44](见Fig.1)。miRNA、降钙素、CRMP2等可能是NaV1.7作用于神经病理性疼痛的关键靶点。
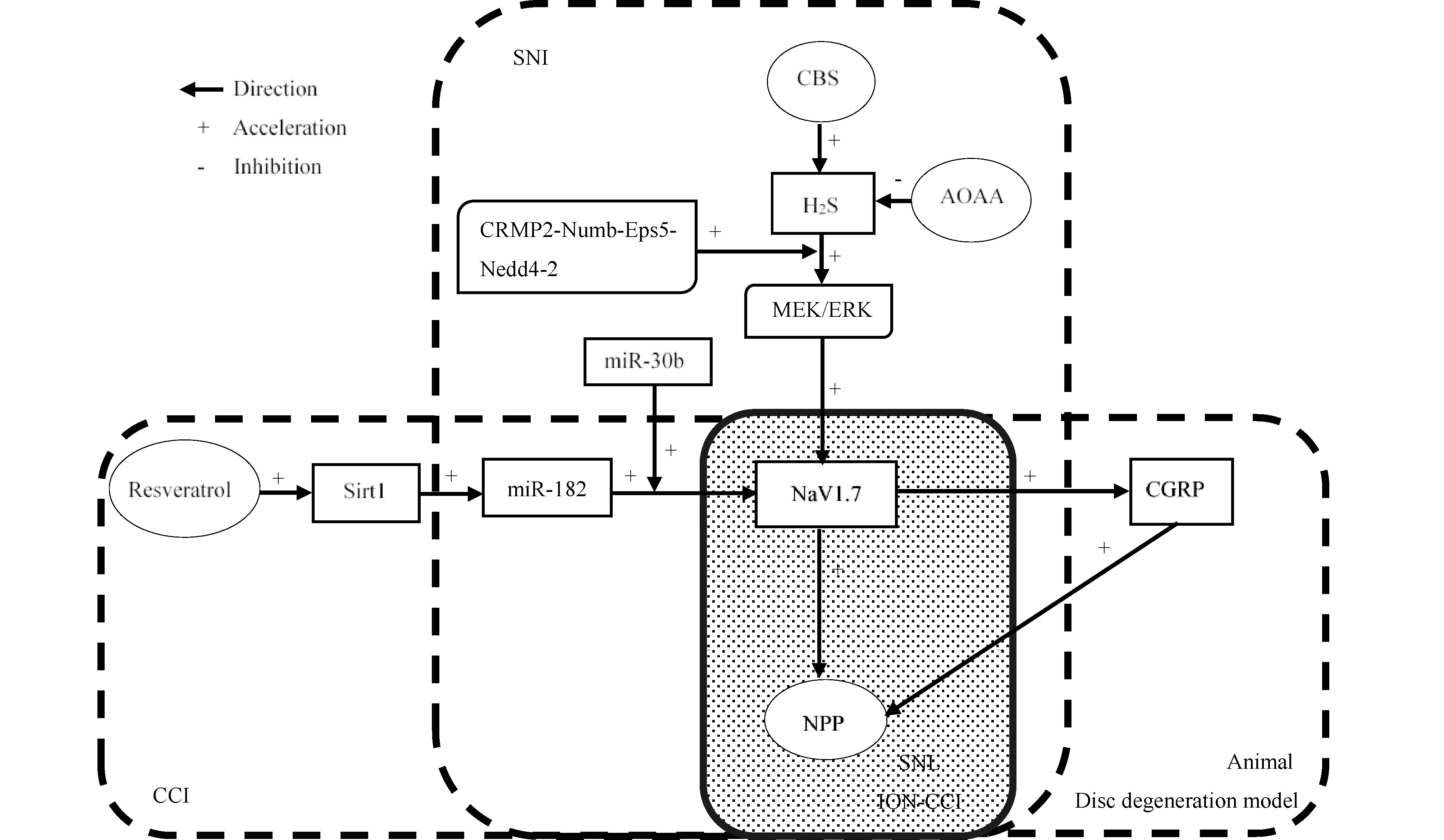
Fig.1 Role of Nav1.7 in neuropathic pain induced by direct nerve injury Nav1.7 is closely related to the abnormal excitability of pain receptors. Nerve injury causes changes in their distribution, expression and / or biophysical properties, which helps to produce and maintain neuropathic pain. MicroRNA, calcitonin and CRMP2 may be the key targets of Nav1.7 on neuropathic pain
2.2 NaV1.7在疾病、药物毒副作用等引起的神经病理性疼痛中的作用
骨癌疼痛是由恶性骨肿瘤侵袭或转移到骨上引起的神经病理性疼痛。将Walker256乳腺癌细胞注射于大鼠胫骨结节,构建大鼠骨癌疼痛模型。模型大鼠DRG神经元中,NaV1.7表达水平随着模型时间的延长而显著升高,骨癌同侧脊髓中星形胶质细胞显著增殖。鞘内注射NaV1.7 短发夹RNA(short hairpin RNA, shRNA)慢病毒载体,NaV1.7表达水平明显恢复,星形胶质细胞特异性标记物胶质原纤维酸性蛋白(glial fibrillary acidic protein, GFAP)上升速度明显减缓,小胶质细胞特异表达补体受体-3的单克隆抗体(clone name of monoclonal antibody of complement receptor type 3, OX42)下降速度明显加快,星形胶质细胞和小胶质细胞的活化受到抑制[45]。粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)在人恶性骨肉瘤中过表达。GM-CSF激活Janus激酶2(Janus kinase2,Jak2)信号转换器和转录蛋白激活因子3(activator of transcription protein 3,Stat3)信号通路,促进DRG神经元中NaV1.7转录,增强神经元兴奋性,产生骨癌相关疼痛[46]。表明NaV1.7在骨癌疼痛模型大鼠DRG神经元中表达升高可能是由于肿瘤压迫局部神经纤维造成损伤,并将伤害性信息持续传递给DRG神经元,刺激DRG神经元细胞持续去极化和激活,促进NaV1.7时间依赖性表达增加所致[45]。
单独使用链脲佐菌素(streptozotocin, STZ)或化疗药物紫杉醇能引起周围神经痛。经STZ诱导大鼠产生糖尿病周围神经痛(diabetic peripheral neuralgia, DPN)后,大鼠DRG和脊髓神经元中NaV1.7表达增加,在长时间内表现出机械异位痛和热痛觉过敏。紫杉醇诱导大鼠神经病变和人神经性疼痛(chemotherapy-induced peripheral neuropathy, CIPN),DRG神经元中NaV1.7表达上调,小直径DRG神经元的瞬时Na+电流和动作电位放电频率增强[47]。与体外研究采用紫杉醇(0.1~1 mol/L)培养原代人背根神经节24 h后NaV1.7表达和放电特性一致[48]。有研究指出,STZ与紫杉醇均能诱导DRG神经元细胞外信号相关激酶1/2(extracellular signal-related kinase 1/2, ERK1/2)磷酸化,活化的磷酸化细胞外信号相关激酶1/2(phospho-extracellular signal-related kinase 1/2,p-ERK1/2)在DRG神经元中与NaV1.7共定位,对NaV1.7通道L1的多个位点进行磷酸化,从而调节NaV1.7通道的门控特性,提高DRG神经元兴奋性[49]。推测CIPN表型[47]产生的原因之一可能是由Toll样受体4(toll-like receptor4,TLR4)-脊髓分化一级反应基因蛋白(myeloid differentiation primary response gene protein 88,MyD88)- pERK1/2信号通路,或/和TLR4-MyD88-P38(phospho-P38,pP38)信号通路联合作用NaV1.7导致[47]。而加巴喷丁(gabapentin, GBP)缓解糖尿病周围神经病理性疼痛大鼠痛觉过敏的镇痛作用,可能是通过NaV1.7与p-ERK1/2相互作用实现的[50]。暗示NaV1.7与p-ERK1/2之间的相互作用为NaV1.7选择性抑制剂研发提供了可能。
除此之外,高血糖的毒性作用[51]激活蛋白激酶C(protein kinase C,PKC),通过NaV1.7通道DⅢ-DⅣ连接体中丝氨酸残基增强了通道的再生电流[52],促使DRG神经元中NaV1.7表达增加[52],引起糖尿病周围神经病变相关神经性疼痛。抑制肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)的合成,阻断核因子-κB(nuclear factor kappa-B,NF-κB)信号通路表达,可明显抑制DRG神经元中NaV1.7的过表达,发挥预防糖尿病周围神经病变的作用[53]。TLR4/NF-κB信号通路的激活同时参与了皮肤/肌肉切开和收缩(skin/muscle incision and retraction, SMIR)诱导的慢性术后疼痛(chronic postoperative pain, CPSP)的发展。在SMIR诱导的慢性术后疼痛中,TLR4/NF-κB信号通路诱导DRG神经元中NaV1.7上调可引起CPSP中的外周痛觉过敏[54]。神经生长因子(nerve growth factor, NGF)通过激活糖皮质激素诱导激酶1(serum and glucocorticoid-inducible kinase 1, SGK1)依赖的Nedd4-2磷酸化,上调NaV1.7(即NGF/trkA-SGK1-Nedd4-2-NaV1.7信号通路)也作用于SMIR诱导的慢性术后疼痛的发展[55]。此外,大鼠足底切口构建急性术后疼痛模型,L4-L6DRG神经元内NaV1.7表达显著上调,鞘内注射NaV1.7干扰慢病毒,可显著抑制DRG神经元中NaV1.7表达,减轻痛觉过敏[56]。因此,NaV1.7也是理解术后疼痛机制的关键。
综上所述,NaV1.7在疾病、药物毒副作用引起的神经病理性疼痛中具有重要作用。胶质细胞的活化、ERK信号通路或NF-κB信号通路的激活以及Nedd4-2等有助于深入了解NaV1.7通道与神经病理性疼痛之间的关系,为NaV1.7选择性抑制剂的研发提供了可行的方案(见Fig.2)。
2.3 NaV1.7在SCN9A基因突变引起的神经病理性疼痛中的作用
NaV1.7通道在痛觉中的作用最初是通过遗传学研究发现的。遗传性红斑肢痛症(inherited erythromelalgia, IEM)是一种神经性疼痛综合征,也是人类神经病理性疼痛的遗传模型[7]。IEM与NaV1.7钠通道编码基因SCN9A的功能获得性突变有关。目前,已有多种SCN9A-IEM相关突变位点被报道(见Table.1),例如F1449V[57]、L858H[58]、L823R[59]、I234T[60]、Q875E[61]、A1632E[62]、V1316A[63]、A1632T[64]以及ΔL955[65]等。在激活NaV1.7钠通道过程中,SCN9A-IEM相关突变均产生了较大的超极化偏移,减慢了从打开状态(失活)到关闭通道的速度,增加了通道保持在打开状态的可能性,增强了NaV1.7通道对小刺激的斜坡电流的反应,使得DRG神经元产生高兴奋性(阈值降低和重复放电增强),从而表现出由温暖或运动引起的阵发性烧灼痛和四肢红斑等IEM特征。但L823R诱导的突变需要引入额外的正电荷才导致明显的超极化偏移,而Q875E突变需要引入额外的负电荷;V1316A突变在NaVβ4肽存在下,将NaV1.7钠通道的失活和活化解耦,从而增强了持续Na+电流产生;ΔL955突变使得NaV1.7通道的持续电流增强,A1632T突变减缓NaV1.7通道快速失活。但两者均未产生再生电流,再生电流似乎对另一种神经性疾病发挥决定性作用;而A1632E突变在不同神经病理性疾病下产生不同的电生理效应。
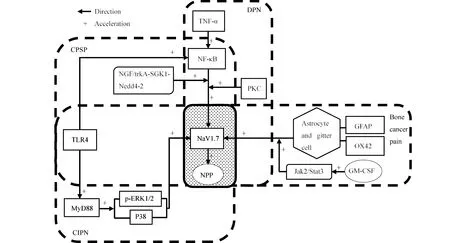
Fig.2 Role of Nav1.7 in neuropathic pain caused by disease, drug toxicity and side effects Nav1.7 plays an important role in neuropathic pain caused by disease and drug toxic and side effects. Glial cell activation, ERK signaling pathway, NF- κB signal pathway or Nedd4-2 helps to deeply understand the relationship between Nav1.7 channel and neuropathic pain, and provides a feasible scheme for the research and development of Nav1.7 selective inhibitors
阵发性极度疼痛(paroxysmal extreme pain disorder,PEPD)是第2种可归因于NaV1.7钠通道α亚基编码基因突变的遗传性疼痛疾病,大约有2/3的PEPD家族存在SCN9A基因突变[57]。SCN9A-PEPD相关突变通过稳态快速失活,减慢开放通道快速失活速度和持续电流的去极化偏移来破坏快速失活。I1464T突变体增加NaV1.7通道内持续电流[66],已被证明增加DRG神经元的钠电流再生[67],T1464I突变体表现出更强的再生电流振幅,两种突变体均表现出不完全失活[57];对于M1627K突变体,更大的去极化偏移和电压依赖性的降低,将扩大“窗口电流”(激活-失活门控重叠)作用的电位范围[57, 68],M1627K突变体增强NaV1.7通道再生电流振幅[67]。V1299F与V1298F突变位于DⅢ S4-S5段,对NaV1.7通道电导的电压依赖性影响很小,主要作用是在去极化方向上改变快速失活的电压依赖性[66];L1612P突变体在电生理特性上显著增加斜坡电流,并缩短了失活恢复时间。但是无持续电流产生,使NaV1.7通道功能增益,提高了细胞高兴奋性[69](见Table1)。
小纤维神经病(small-fibre neuropathy,SFN)在许多罕见的遗传疾病中都有表现,其特征为远端无髓鞘/薄髓鞘纤维丢失,同时产生感觉功能障碍和神经性疼痛[70]。在约30%的特发性SFN(idiopathic SFN,ISFN)病人中发现,存在SCN9A基因功能获得变异体[8]。钠通道NaV1.7的功能获得变异改变了钠通道快速失活、缓慢失活或再生电流,每种突变都使DRG神经元高兴奋性[71]。转染M932L/V991L (ML/VL)和I720K突变体的DRG神经元表现出神经突长度减少的趋势[72];I739V突变体在DRG神经元中的表达使得DRG神经元细胞过度兴奋,降低电流阈值,增加由分级阈上刺激引起的放电频率[73];D623 N突变体使DRG神经元静息电位去极化,并在脉冲间隙产生增强的内流电流,从而提高DRG神经元兴奋性[74];体外观察转染I228 M突变体的DRG神经元表明,DRG神经元的神经突长度减少,功能获得性突变的NaV1.7表达可破坏线粒体,并损害细胞产生ATP的能力,由此产生的生物能量危机会导致SFN中轴突的丢失[72, 75],而敲入NaV1.7-I228 M突变体的小鼠细胞系再现了与NaV1.7钠通道的功能获得突变相关的DRG神经元高兴奋性,但无机械或热痛觉过敏或皮内神经纤维丢失,即在病人中所见的疼痛或神经病变表型[70](见Table1)。

糖尿病周围神经病变(diabetic peripheral neuropathy, DPN)是糖尿病常见的致残并发症[51]。在糖尿病周围神经病变中,糖尿病引起的自主神经轴突的结构性损伤对自主神经功能产生重大影响,而NaV1.7突变对自主神经元兴奋性无直接影响[76],NaV1.7通道的编码基因突变可能与胰腺β细胞衰竭和DPN的开始有关[77]。具有遗传易感性基因的患者体内易感环境促进NaV1.7突变的产生,导致钠通道功能获得性增益,引起胰腺β细胞病变,并随后引发糖尿病和DRG神经元的高兴奋性[9]。表明SCN9A突变可能与糖尿病周围神经病变相关的神经性疼痛和疼痛严重程度有关[78]。
SCN9A基因的双等位基因功能缺失突变导致先天性疼痛不敏感(channelopathy-associated insensitivity to pain,CIP)产生,CIP患者无法感受到伤害性(机械、热或化学)刺激引起的疼痛。NaV1.7基因敲除小鼠[79]或在感觉和交感神经元中删除SCN9A基因[3],均能成功再现了人类先天性疼痛不敏感的表型。SCN9A-CIP相关基因突变导致NaV1.7蛋白功能失调或不产生相应蛋白质[80],但部分突变位点仍保留NaV1.7通道功能(见Table1),例如W1775R和L1831X突变体在NaV1.7通道激活中表现出去极化偏移、A1236E和L1831X突变导致稳态快速失活的超极化偏移[81]、NaV1.7-R896Q突变使得NaV1.7通道蛋白错误折叠,从而在内质网滞留,导致NaV1.7的细胞表面定位缺陷等[82]。MMP-9属于基质金属蛋白酶(matrix metalloproteinase,MMP)家族,主要负责降解和重塑细胞外基质的动态平衡。CIP-R896Q突变序列与预测的MMP-9切割位点序列重叠,对MMP-9蛋白水解具有较高的敏感性。在神经发生过程中,MMP-9蛋白水解异常加速可能是NaV1.7失活的生化基础,突变体的增强裂解可能是造成CIP的原因之一[83]。因此,SCN9A基因突变如何通过NaV1.7变化导致CIP还有待深入了解[84]。
3 NaV1.7的靶向药物研究现状
NaV1.7选择性抑制剂的研发是生物医学研究和学术界广泛关注的热点和难点。根据NaV1.7通道的结构特点,VSDⅣ、VSDⅡ和通道孔的细胞外前庭为研发NaV1.7选择性抑制剂提供了可能。(1)PF-05089771[85]、PF-04856264[86]、GX-674[16]、GX-936[16, 87]、AMG8379[88]等磺胺类药物与VSDⅣ结合[16, 89],具有较强的“状态依赖性”[90]。PF-05089771[85]耐受性良好,但在临床试验中疗效不明显。在游离血浆蛋白浓度较低情况下,PF-06456384无法在福尔马林诱导的疼痛模型中发挥抑制作用[86]。目前磺胺类药物在NaV亚型选择性和靶向性方面的研究已得到显著改善,但这些改进能否安全有效地在人类疼痛中发挥有力镇痛作用仍有待证明[14]。(2)胱氨酸结肽与VSDⅡ结合[14]。Pn3a[91]、ProTx-Ⅱ[92, 93]、GPTX1[94, 95]等相关抑制剂具有明显的NaV1.7选择性、抑制效力和靶向作用,但Pn3a[91, 96]与ProTx-Ⅱ[93]在啮齿类动物急性伤害性或炎性疼痛中未见表现出镇痛作用,GPT1-mAb(GPTX1衍生物通过聚乙二醇的接头位点特异性结合到非靶向单克隆抗体mAb上),虽能显著影响体外抗NaV1.7的效力,但不能改变组胺诱导的瘙痒模型中的抓挠行为,在其他疼痛模型中的研究报道较少[97]。(3)胍类化合物与通道孔的细胞外前庭结合。河豚毒素和石房蛤毒素(saxitoxin, STX)是一类天然存在的钠通道抑制剂,通过与细胞外前庭结合并堵塞通道孔,“状态依赖性”低[98, 99]。NaV通道亚型之间存在很小的序列差异,但能显著影响配体效力[99]。由于NaV通道亚型之间具有高度保守的性质,这类胍类选择性抑制剂研发存在困难。随着研究的不断深入,抗NaV1.7单克隆抗体产生,例如SVmab和SVmab1等[100]。SVmab、SVmab1作用于VSDⅡ的S3-S4连接子,通过稳定通道的失活状态来阻止通道激活,从而将激活的电压依赖性转移到更去极化的状态[100, 101],在炎症、急性或慢性瘙痒以及神经病理性疼痛中抑制NaV1.7表达[101]。但离子通道在短时间内(毫秒或更少)发生多种与门控相关的构象变化,使得抗体无法与NaV1.7的各构象异构体实现快速高效结合,暗示筛选高亲和力抗体结合作为NaV1.7选择性抑制剂的首选方案并不理想[100]。因此,充分了解NaV1.7通道及其在不同因素引起的神经病疼痛中的作用机制至关重要。
4 问题与展望
神经病理性疼痛是慢性疼痛中最复杂多变的疼痛类型之一。目前,广泛用于治疗神经病理性疼痛的方法,包括抗抑郁药、抗惊厥药和镇痛药等,都具有一定的中枢神经系统副作用,例如镇静和协调障碍等[102]。而NaV1.7选择性抑制剂存在选择性、效力、靶向性、安全性以及可行性等多方面问题[11, 14]。因此,充分了解NaV1.7与其他NaV通道亚型之间的序列差异、通道结构特点、生物物理学性质以及NaV1.7在不同神经病理性疼痛中的作用机制,是研发选择性更强、毒副作用更小、镇痛效果更明显的新型NaV1.7选择性抑制剂亟待解决的首要问题。寻找NaV1.7作用于不同神经病理性疼痛的普遍机制,或NaV1.7特有的受体结合位点可能是未来NaV1.7选择性抑制剂研发的方向。
