SEMA6B 基因变异致进行性肌阵挛性癫痫1 例报告
2022-09-01赵金华汤继宏张兵兵邢玉娇师晓燕
赵金华 汤继宏 黄 静 肖 潇 张兵兵 邢玉娇 师晓燕
1.苏州大学附属儿童医院神经内科(江苏苏州 215025);2.南通市第一人民医院儿科(江苏南通 226001)
进行性肌阵挛性癫痫(progressive myoclonic epilepsy,PME)是一群罕见的临床和基因异质疾病,其特征在于肌阵挛、癫痫和神经系统恶化。神经系统恶化可能包括神经病变、共济失调、渐进性认知退化和肌病[1]。大多数疾病以常染色体隐性遗传方式遗传,少数表现为常染色体显性遗传或线粒体遗传。随着分子遗传学技术的发展,已经发现许多基因(如GOSR2、ASAH1、KCTD7、TBC1D24、SCARB2、Spirkle 1、CARS 2、SERPINI)的变异可导致PME。PubMed检索非常有限的病例报告表明,SEMA6B的致病变异也可引起PME 11型(OMIM:618876)。本文回顾分析1 例SEMA 6 B基因变异所致PME 11 型患儿的临床资料及全外显子组测序结果,以提高对SEMA6B基因变异相关癫痫疾病的认识。
1 病例资料
患儿,女,15岁,汉族,因“发育迟缓14年,运动及认知明显倒退1 年余”于2021 年3 月入住苏州大学附属儿童医院。患儿生后18 个月独走,运动发育较同龄儿稍落后,语言、智力发育也落后,但能跟同龄儿一起玩耍及交流。入院前1 年多开始四肢抖动明显,双手持物不稳,步态不稳、宽步,易摔倒,逐渐丧失独立行走能力,行走困难,外出时需坐轮椅。时有不自主肢体抽动、耸肩及躯体扭动。能回答简单问题,但思维迟缓,构音障碍,言语不清,计算能力倒退。患儿6岁时出现第一次抽搐,表现为全面性强直-阵挛性发作,后又反复发作,在当地医院诊断为癫痫,予丙戊酸钠抗癫痫治疗7年,期间无抽搐发作,但时有不自主肢体抖动,意向性震颤。2年前考虑丙戊酸钠相关副作用,换用左乙拉西坦抗癫痫治疗,但效果欠佳,仍然反复出现身体抖动,甚至偶尔出现全面性强直-阵挛性发作,每次约2~4 min。1年前自行停用抗癫痫药物,期间仍有反复抽搐发作。
患儿系G2P2,足月顺产,出生体重3 400 g,出生时有脐带绕颈、轻度窒息,Apgar 评分不详。父母体健,非近亲结婚。无家族遗传病史。
入院查体:体温37 度,脉搏78 次/min,呼吸18次/min,身高170 cm,体重48 kg。头围正常。神志清,言语不清,讲话断断续续,记忆力差,计算力差,简单加减法已不能计算。双侧瞳孔等大等圆,颈软,呼吸平稳,两肺呼吸音粗,未闻及啰音,心律齐,未闻及杂音。腹软,肝、脾肋下未触及,四肢肌力5级,肌张力增高,膝反射亢进,共济失调,病理征未引出。入院后辅助检查:血常规、C 反应蛋白、凝血功能、血氨、血培养、血沉、输血前检查、体液免疫、乙肝三对、生化全套、脑脊液常规、生化、培养等均正常。心电图未见异常,血、尿遗传代谢筛查正常。头颅MRI颅内未见明显异常。长程视频脑电图检查:监测过程中数次临床发作,表现为双上肢抖动一下,同期及发作间期脑电图示醒睡各期均可见额颞部为主的全导中-高波幅2~3Hz棘慢波、多棘慢波频繁阵发发放,睡眠期痫性放电明显减少;也可见睡眠期双侧枕部和中颞部中-高波幅2~3Hz棘慢波阵发发放,右侧枕、中颞部显著(图1)。
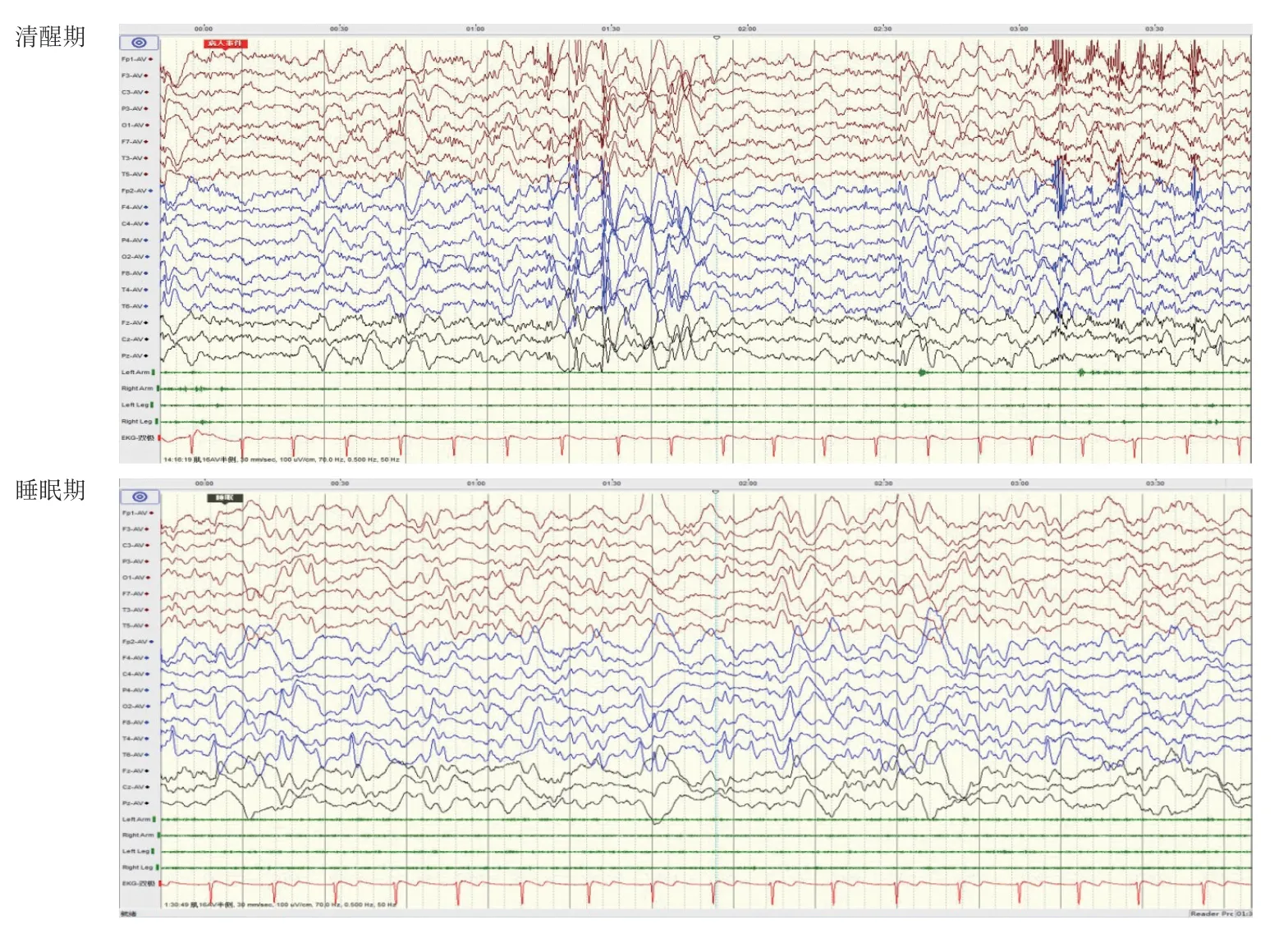
图1 患儿视频脑电图
入院后经基因检测发现,SEMA 6 B基因有一无义变异:c.2149(exon17)C>T,导致氨基酸改变p.Q717*,172(p.Gln717Ter,172);根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南,该变异为可能致病。具体依据为:PS2+PM2+PM4,即强致病证据PS 2:该变异在经亲缘关系确定的家系中为新发变异;中等致病证据PM 2:所有正常人群数据库频率小于0.0005;中等致病证据PM4:非重复区框内插入/缺失导致的蛋白质长度变化。目前尚未见该变异文献报道;家系验证发现其父母该位点均未见基因变异,该变异为新生变异(de novo),结合临床表型确诊为PME 11型(OMIM:618876),该疾病为常染色体显性遗传。对SEMA6B基因的疑似变异用Sanger法进行验证,其结果与二代测序一致(图2)。患儿诊断治疗时间轴见表1。

表1 患儿诊断及治疗时间轴

图2 SEMA6B 基因疑似变异的Sanger 测序图
2 讨论
以“SEMA 6 B”为关键词,分别在中国知网和万方数据库进行检索,自建库至2021 年5 月,未见相关中文文献。在PubMed数据库中进行检索,自建库至2021 年5 月,国际上仅2 篇文献报道SEMA 6 B基因变异患者,共6 例[2-3]。对6 例文献报道的患者及本研究1 例患儿的临床资料进行总结分析。7 例患者均为散发患者,起病年龄为11 月~6 岁,首发症状均为癫痫发作,有4 例发作类型为全面强直阵挛发作,1 例发作类型多样,还有2 例发作类型不详。7 例患者均有小脑共济失调、震颤发作,6 例伴智力运动发育落后,有3 例伴小脑畸形。头颅MRI发现1 例小脑萎缩、1 例小脑蚓体偏小,其他5 例正常。7例患者均为新生变异(de novo),有2例变异位点相同。见表2。
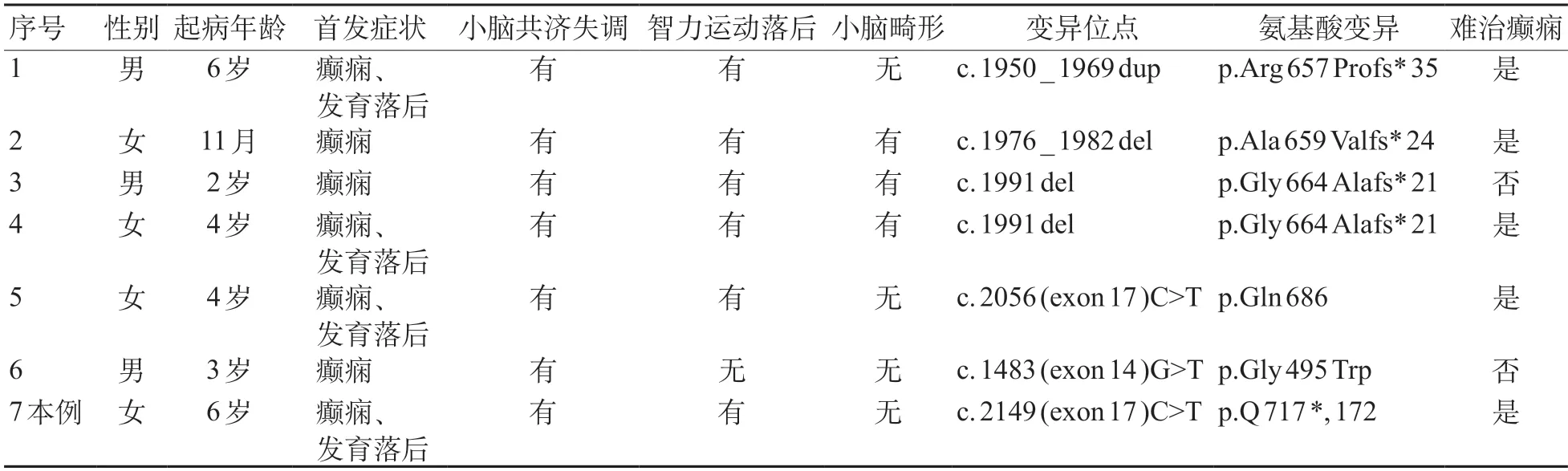
表2 SEMA6B基因变异7例患者临床特点和基因检测结果
PME是一种罕见的异质性遗传疾病,具有肌阵挛、癫痫发作和进行性神经功能恶化的三联征,占所有癫痫不到1%,多数为常染色体隐性遗传,但也有罕见的常染色体显性和线粒体遗传类型[4]。PME的病因包括一组神经遗传病,包括如神经元蜡样质脂褐质沉积症(NCL)、Lafora病、唾液酸沉积症等十余种疾病,该组疾病通常在儿童或青少年期起病,少数也可在婴幼儿期起病,大多预后不良[1,5]。如NCL婴儿型常在1岁内以智力、运动发育落后起病,随后出现肌阵挛发作和其他形式的癫痫发作,病情持续恶化,多数患儿在10岁前死亡[6]。Lafora病在8~18岁以全面强直-阵挛发作起病,随后很快进展为痴呆伴共济失调以及视力下降,最终在发病后10年内死亡[7]。由于PME非单一病因且临床表现复杂多样,因此病因诊断需要结合发病年龄、临床表现、头颅影像学、视听功能评估,酶学检测和皮肤活检等,但仍有部分患儿病因诊断困难[8]。近年来随着二代测序技术在临床上的应用,新发现了多种基因变异可导致PME表型,如ASAH1、KCNC1、GOSR2、KCTD7、TBClD24、SCARB2、PRICKLEl、CARS2和SERPINll基因等。本研究报道的SEMA 6 B基因变异是一种新发变异,2020 年4 月发表的文献首先总结了该基因变异相关性PME,并认为其NMD(nonsensemediated mRNA decay)(α)区域中的截断DNV(de novo variants)导致PME[2]。SEMA6B在不同发育阶段的不同大脑区域的广泛表达,在不同层次的兴奋神经元中表达,在大脑发育过程中参与γ-氨基丁酸(GABA)能的神经元间迁移,在GABA能神经元中SEMA 6 B 功能的破坏可能导致人类癫痫。SEMA6B在神经系统障碍发病机制中的重要作用,需要在体内或体外SEMA 6 B 模型中进行进一步的工作来阐明SEMA6B细胞内结构域截断在PME中的功能机制[2]。
Song 等[3]近期研究表明SEMA 6 B 通过介导Semaphorin/Plexin信号传导引起癫痫,对SEMA6B和PlxnA 2 相互作用的进一步研究支持了无义变异导致SEMA 6 B 和PlxnA 2 蛋白相互作用增加的事实,无义变异引起的有害截断蛋白显著地增加了与PlxnA 2 的结合,最终导致无义变异比错义变异更严重的临床表现。本例患儿发现SEMA6B基因亦是无义变异,为新发变异且截短变异在外显子17,与Song等[3]报道的第一例患儿相似,为极强致病证据(PVS1)。本例患儿临床表现为肌阵挛,意向性震颤,共济失调,逐渐丧失独立行走能力,行走困难,外出需坐轮椅,腱反射亢进,肌张力增高,构音障碍,言语倒退,智力障碍,多灶性癫痫发作,脑电图示典型痫性放电,癫痫难治等,其基因变异致病等级为PVS 1+PS 2+PM 2+PM 4,提示本例患儿SEMA 6 B基因无义变异的致病性明确,危害性严重,与患儿表型相符。
本研究及文献6例SEMA6B基因变异患者临床表型主要表现癫痫发作、小脑共济失调、震颤发作、伴智力运动发育落后,可伴小脑畸形,符合PME 临床表现。但7 例SEMA 6 B基因变异患者病情相对稳定,年龄最大者已28岁,未发现早期死亡患者,与其他疾病导致的PME表型相比较轻。文献报道的6例SEMA6B基因变异所致PME患者均有小脑共济失调、震颤发作等表现,半数有小脑畸形,提示小脑畸形在SEMA6B基因变异患者中并非罕见。本研究患者头颅MRI未见小脑畸形,但其已有小脑共济失调及震颤表现,因此需继续随访。
PME的治疗基本上是抗癫痫发作和肌阵挛,以及姑息、支持和康复措施。对治疗的反应最初是相对良好的,之后,癫痫发作可能变得更频繁,并且发生逐渐出现的神经系统功能下降[1]。传统的抗癫痫药物,如丙戊酸钠、左乙拉西坦、苯二氮卓类等,这是肌阵挛和癫痫发作的初始治疗方法,然而,单一治疗和不同药物联合治疗通常无法控制所有的致残临床症状,而生酮饮食对PME 相对不成功,有前途的治疗方法似乎来自神经调节领域。迷走神经刺激术(VNS)和深部脑刺激(DBS)都证明有效,对耐药患者能减少癫痫发作和/或肌阵挛发作。酶替代治疗和基因治疗被证明是有效的精确疗法,可能改变疾病过程,因此是主要期望[9]。
本病例开始未行基因检测,确诊时间较晚,因此对于此类患儿,需强调早期基因检测的重要性。由于患儿家属依从性较差,不配合治疗,故此患儿预后不良。
本例患儿基因变异 c.2149(exon17)C>T为新生变异,查阅既往文献及数据库未见该位点的相关性报道,该基因变异位点的发现扩充了PME的基因变异谱。
