真核翻译起始因子2α激酶在肾脏疾病中的作用研究进展*
2022-08-20郑笑慈
郑笑慈 吴 嵽 徐 昕
(上海体育学院,上海兴奋剂检测实验室,上海 200438)
据统计,全球约有8.5亿人患有肾脏疾病,包括慢性肾脏病(chronic kidney disease,CKD)、急性肾损伤(acute kidney injury,AKI)和肾衰竭(renal failure,RF)等[1]。随着患者肾脏功能的衰退,死亡风险呈指数级升高且伴有高致残率,易导致患者的生活质量水平显著降低,昂贵的治疗费用也会给患者及其家庭带来巨大的经济负担[2-3]。因此,肾脏疾病的治疗亟需学者进一步的研究并获得新的治疗靶点。
应激反应机制通过多层调节控制细胞命运。翻译的衰减是由各种压力触发的快速细胞反应,如整合应激反应(integrated stress response,ISR)和mTOR抑制[4]。ISR是一种进化保守的信号级联反应,指在多种压力刺激下,通过磷酸化真核翻译起始因子2α(eukaryotic translation initiation factor 2α,eIF2α)激酶所介导的细胞适应反应[5]。eIF2α激酶家族包括一般性调控阻遏蛋白激酶2(general control nonderepressible 2,GCN2)、蛋 白 激 酶RNA样内质网激酶(protein kinase RNA-like endoplasmic reticulum kinase,PERK)、双链RNA依赖性蛋白激酶(double stranded RNA activated protein kinase,PKR)和血红素调节抑制蛋白激酶(heme-regulated inhibitor kinase,HRI)[6]。eIF2α的磷酸化使其与鸟嘌呤核苷酸交换因子eIF2β的结合更加紧密,阻止小核糖体亚基的形成,导致翻译减弱[7]。
eIF2α激酶是哺乳动物细胞中代谢应激反应的关键因子,可诱导翻译的整体停滞,恢复蛋白质合成,控制细胞存活。eIF2α激酶的失调与多种人类疾病的病理生理学有关,如糖尿病[7]、癌症[6]、病毒感染[8]和神经退行性疾病[9]等。研究提示,eIF2α激酶可能参与多种肾脏疾病的病理生理过程。因此,本文对eIF2α激酶家族及其在肾脏疾病中的可能作用等方面的研究进展进行归纳总结,以期为肾脏疾病的防治提供新的参考和依据。
1 eIF2α激酶家族简介
1.1 GCN2
GCN2(或称eIF2αK4)是一种丝氨酸/苏氨酸蛋白激酶,最初在芽殖酵母中发现,负责检测和感应酵母中氨基酸缺乏[10]。丝氨酸/苏氨酸蛋白激酶是细胞质结构域的一部分,负责与高催化活性相关的受体活化。GCN2编码1 649个氨基酸(amino acid,AA),分子质量约为190 ku(二聚体为380 ku)。GCN2有5个折叠结构域:N端RWD(指环蛋白,含WD重复序列的蛋白质和酵母DEAD样解旋酶)结构域、假激酶结构域(pseudokinase domain,PKD)、激酶域(kinase domain,KD)、组氨酸-tRNA合成酶样(homology to histidyltRNA synthetases,HisRS)样结构域、C端二聚化结 构 域(C-terminal dimerization domain,CTD)(图1)[6]。在氨基酸缺乏期间,不带电荷的游离tRNA会增加,GCN2可以感测和结合这些tRNA,通过HisRS样结构域和C端激酶结构域,导致其催化结构域的激活和eIF2α亚基第51位的丝氨酸磷酸化,还可特异性促进某些基因mRNA的翻译,从而抑制整体mRNA的翻译及蛋白质的合成[11]。GCN2参与数百个基因的转录和表达,对维持细胞稳态、参与蛋白质代谢、免疫反应、炎症反应及其他生理和病理反应均有重要作用。
1.2 PERK
PERK(或称PEK,eIF2αK3)最初是在小鼠的研究中发现的[12]。PERK是位于内质网膜中的一种跨膜蛋白,编码1 116个AA,分子质量约为125 ku,它具有两个不同的结构域:N端区域位于内质网腔内,包含免疫球蛋白重链结合蛋白(immunoglobulin heavy chain binding protein,BiP)和热休克蛋白5(heat shock protein family A(Hsp70)member 5,HspA5),是调节和结合蛋白质的重要区域;C端区域是激酶结构域和自磷酸化位点(图1)[13]。PERK被内质网中错误折叠的蛋白质积累所激活,通过磷酸化eIF2α,可以阻止新生多肽的合成,减少新生多肽进入内质网。PERK主要分布在分泌型组织中,其中在胰腺中表达量最高,在其他组织中也有少量分布,其主要功能是负责内质网应激信号的传导,参与内质网相关蛋白质的翻译调节[12]。
1.3 PKR
20世纪90年代初,PKR(或称eIF2αK2)在干扰素(interferon,IFN)处理的病毒感染L细胞中首次被发现,其双链RNA依赖的激酶活性早为人知[14-15]。PKR是一种编码551个AA、分子质量约为68 ku的丝氨酸/苏氨酸激酶,存在于细胞质和细胞核中,具有两种不同的结构域,包括N端调节的双链RNA结合结构域和C端激酶结构域(图1)[16]。PKR是一种关键的抗病毒蛋白,同时也是先天免疫的重要组成部分,在IFN对抗和获得性免疫反应开始之前的宿主防御中发挥作用[17]。除了细胞、病毒或合成来源的dsRNA外,PKR还可以被Toll样受体、生长因子受体和细胞因子(例如白介素1和肿瘤坏死因子α)以及各种细胞应激诱导剂(如亚砷酸盐、过氧化氢等)激活[18]。此外,在应激条件下,由病毒或非病毒刺激激活的PKR激活蛋白(PKR-activating protein,PACT)可以作为一种介质,以直接的蛋白质-蛋白质相互作用方式激活PKR[19]。PKR与dsRNA结合后,经过二聚化和自磷酸化,磷酸化其底物,包括eIF2α、蛋白磷酸酶2A和IκB激酶,通过这些底物和下游效应物,PKR可以调节细胞翻译、转录、凋亡和细胞分化等[18]。
1.4 HRI
HRI(或称eIF2αK1)是一种主要在红细胞和偶联蛋白合成中表达的蛋白激酶[20]。最初认为HRI表达仅限于红细胞,主要在红系细胞中表达[21],但有研究表明,它也在肝脏和巨噬细胞中表达[22]。HRI编码630个AA,分子质量约为71 ku。HRI有5个结构域:N端、激酶Ⅰ结构域、激酶插入(kinase insert,KI)结构域、激酶Ⅱ结构域、C端(图1),其中N端、KI端和C端是HRI特有的,血红素结合位点存在于N端和KI端。N端包含稳定的血红素结合点,这是血红素调节HRI所必需的。而KI端包含可逆的血红素结合位点,血红素通过侧接组氨酸残基在两个位点进行协调[20]。HRI作为一种血红素调节的翻译抑制剂,通过感知细胞内血红素浓度平衡血红素和珠蛋白的合成。当血红素浓度较低时,HRI被自磷酸化激活,在其N端与血红素结合,一旦结合会触发HRI分子间自磷酸化,从而稳定HRI的聚集,并产生能够感知细胞内血红素浓度的HRI二聚体;当血红素浓度较高时,血红素与HRI结合,导致HRI激活被抑制,从而合成珠蛋白;当血红素缺乏时,HRI被自磷酸化激活,激活的HRI进一步磷酸化eIF2α,抑制珠蛋白的合成[23]。
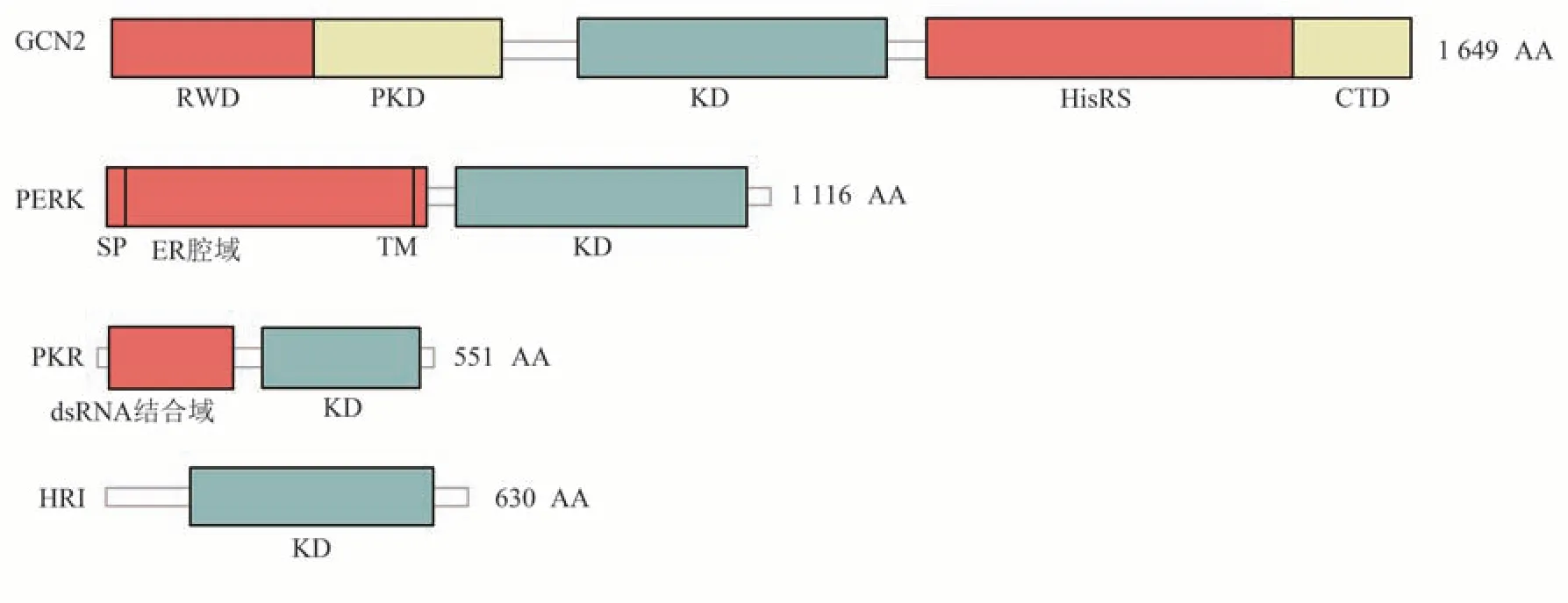
Fig.1 Domain organization of the eIF2αkinases family图1 eIF2α激酶家族的结构域
2 eIF2α激酶家族在肾脏疾病中的作用
2.1 GCN2
GCN2激酶可以直接检测到细胞内氨基酸的缺乏,它结合了不带电荷的tRNA[11],因此,理论上GCN2激酶可检测到任何氨基酸或非必需氨基酸中的缺乏。GCN2可使eIF2α磷酸化,抑制起始复合物的形成,从而抑制一般翻译的开始[6]。同时,当翻译启动效率低时,全长开放阅读框(open reading frame,ORF)上游的微型ORF被跳过时,翻译效率低下的在非编码微-ORF下游具有起始密码子的特定mRNA会被抑制[24]。驱动对氨基酸缺乏有转录反应的转录因子,例如激活转录因子4(activating transcription factor 4,ATF4)[25]。在ISR中,ATF4具有调节正常代谢和氧化还原过程的重要作用。此外,ATF4在调节肥胖、葡萄糖稳态、能量消耗和神经可塑性等方面发挥着核心作用[7]。GCN2信号对细胞表型具有广泛的影响,可以调节与氨基酸的合成和转运,调节氧化还原反应、脂肪酸产生和与炎症相关基因的表达[26]。在哺乳动物中,氨基酸除了被GCN2信号途径感知以外,还可被mTOR信号途径感知。有研究表明,GCN2-eIF2α和mTOR通路之间存在重要串扰,两者可能相互作用调节[25]。mTOR信号强烈感知特定氨基酸的存在,主要是亮氨酸,其次是精氨酸和蛋氨酸[26]。mTOR信号传导途径整合营养、能量和生长因子的可用性,促进合成代谢过程(如脂肪酸合成),抑制分解代谢过程(如自噬)。
GCN2激酶由于氨基酸限制而被激活[27]。饮食中的蛋白质或氨基酸限制减轻了临床和实验性肾小球肾炎和肾脏缺血的病理变化[28-29]。然而,蛋白质或氨基酸限制在肾脏疾病的病理生理学中的作用尚不明确。在肾脏缺血再灌注的临床前哺乳动物模型中,短期饮食限制可通过多种饮食预处理方案来实现,包括禁食、减少食物摄入、蛋白质限制、氨基酸限制和仅含糖流质饮食等。其中,在单一必需氨基酸色氨酸缺乏的情况下,激活GCN2激酶可使小鼠肾脏免受缺血损伤,诱导炎症减少,有保护肾脏功能的作用[29]。但是,在没有任何饮食干预的情况下,GCN2的缺乏对缺血再灌注起到一定的保护作用。造成以上研究结果不一致的原因可能有以下几种。a.由于GCN2在急性应激前后不同的作用所致。应激前GCN2的激活导致了有益的全身性改变,如:循环白细胞减少、IGF1减少;但在应激后,GCN2的激活可能产生损害,如细胞凋亡信号增加、细胞死亡增多。GCN2的缺乏可能会出于不同的原因而起到保护作用[30]。b.缺少GCN2会导致eIF2α激酶通过GCN2以外的激酶对GCN2信号传导的各个方面进行代偿性激活。例如在GCN2缺失的情况下,mTOR的抑制可能会增加蛋白质合成,增加内质网应激(endoplasmic reticulum stress,ERS),激活未折叠的蛋白质反应,从而激活PERK,稳定ATF4。c.此和GCN2本身的结构有关。在不同的配体结合情况下,GCN2激酶结构域会形成不同的构象状态[6]。这种不稳定的性质会导致GCN2活性的复杂调控,所以需要进行进一步的研究,尤其是对GCN2结构进行分析,以及对酶活性进行动力学分析。除此之外,有研究指出,肾脏自身存在的GCN2对氨基酸的感应并没有直接起到保护作用,并且蛋白质限制的功能益处也不需要GCN2的参与[31]。GCN2未能激活的原因可能是肾脏对肌肉存储释放的游离氨基酸的重吸收增加引起的[32]。由于GCN2在肾脏疾病中作用的两面性,因此,亟待更多的研究进一步阐明。
GCN2的激活会调节ATF4-CHOP信号通路中相关蛋白质及其mRNA的表达,导致细胞周期停滞[33]。肾小管周期停滞与肾小管衰老有关,会导致慢性肾病的进展[34]。有研究发现,较高浓度的D-丝氨酸会导致细胞周期停滞、细胞衰老,同时伴随细胞凋亡的增强,会诱导肾小管细胞毒性,上调促纤维化因子,可能加速CKD进展和肾脏衰老[35]。CKD发病机制可能与D-丝氨酸激活GCN2,诱导下游ISR信号通路,在mRNA水平上调ATF4和CHOP有关[35]。此外,L-丝氨酸给药会改善D-丝氨酸诱导的肾损伤,D-丝氨酸的暴露会引起L-丝氨酸合成的代偿。因此,降低D-丝氨酸的绝对浓度,为CKD患者提供L-丝氨酸,阻断ISR的下游效应,可能会作为CKD的潜在治疗策略。
GCN2磷酸化eIF2α激酶,调节肾髓细胞抵抗高尿素诱导的高渗胁迫能力[36]。在哺乳动物的肾脏中,肾髓质细胞经常暴露于含有波动浓度的尿素和氯化钠溶质中,其中钠和尿素会导致髓质内高渗,而髓质乳头顶端渗透压升高是肾脏尿浓缩功能所必需的[37]。长期以来,肾髓细胞如何在高渗环境下存活并发挥正常功能是研究的热点问题。大多数对高尿素胁迫的研究集中在DNA损伤、细胞周期停滞和氧化应激等方面,关于尿素对细胞存活产生的作用信号通路的研究甚少。其中有研究表明,尿素诱导的GCN2激活增强eIF2α的磷酸化,激活下游转录因子3(ATF3)的增加[36]。ATF3是一种转录调节因子,参与多种生理和病理事件,与组织中的应激反应有关,包括DNA损伤、氧化应激、细胞损伤和致癌刺激物等,其中在肾脏中,ATF3的产生对肾小管细胞起保护作用[38]。GCN2的缺乏使细胞对尿素应激敏感,减少了尿素诱导的eIF2α磷酸化,降低ATF3的表达,上调caspase-3的表达,降低细胞存活率[36]。因此,GCN2的活化有助于肾髓质集合管上皮细胞耐受高尿素诱导的渗透压胁迫,从而维持内环境的稳定。
2.2 PERK
已知ERS在人类各种肾脏疾病的发病机理中起重要作用,也是肾小管细胞中响应有害刺激的关键机制之一[39]。内质网负责正确地修饰和折叠蛋白质,当存在各种内源性干扰或外部刺激时,会导致内腔错误折叠蛋白的积累,引起ERS并诱导下游调节,这种反应被称为未折叠蛋白反应(unfolded protein response,UPR)[40]。ERS是一种保护过程,可增强未折叠蛋白质的降解,提高正确折叠的效率,以维持蛋白质体内平衡。然而,在缺血、感染、缺氧或活性氧超负荷等病理损伤情况下,持续的ERS会导致内质网功能障碍和UPR激活[40]。ERS和蛋白质错误折叠的作用在各种肾脏疾病中都有体现,包括原发性肾小球肾炎、糖尿病肾病、与基因突变相关的肾小球疾病、急性肾损伤、慢性肾病等[40]。大量数据证明,ERS是肾小管上皮细胞萎缩和间质纤维化的关键因素[41]。在肾脏疾病患者的肾脏活检样本中,发现包括XBP1和HspA5在内的ERS标志物显著增加[42]。
哺乳动物细胞中有3种跨膜内质网应激信号分子,负责调节UPR途径,包括激活转录因子6(ATF6)、肌醇需求激酶1(inositol requiring enzyme 1,IRE1)和PERK[43]。在生理条件下,这些信号分子与BiP结合,但受到刺激后,会发生解离,激活下游信号传导,包括抑制翻译、诱导细胞凋亡以及与内质网相关的降解[44]。研究发现,在缺血再灌注脑中,eIF2α磷酸化归因于内质网eIF2α激酶PERK的激活[45]。在缺血再灌注之后,脑和肾脏之间的细胞应激反应得以保留。因此,在进一步缺血性再灌注肾脏eIF2α磷酸化和PERK活化的研究中,发现缺血再灌注肾脏的eIF2α增加了约20倍,并伴有PERK激活。此外,免疫组织化学分析发现eIF2α定位于肾小管上皮细胞,表明这些细胞在肾缺血再灌注期间发生ERS[46]。这些结果表明,PERK可能为研究缺血再灌注后肾脏ERS和UPR表达的意义提供了新的思路。
研究表明,ERS通过调节PERK-ATF4-CHOP通路介导细胞凋亡[47]。PERK通过磷酸化eIF2α,导致ATF4通过独立于eIF2α的途径进行翻译,ATF4易位至细胞核并诱导减轻ERS所需的基因转录,CHOP是ATF4的关键下游靶标,可在内质网应激期间触发凋亡[48]。除此之外,ERS和细胞凋亡之间存在分子串扰机制,特别是在PERK-ATF4-CHOP信号通路上。研究发现,过量的铜可能通过激活PERK-ATF4-CHOP信号通路来诱导ERS,随后,ERS会加重铜诱导的肾小管上皮细胞凋亡并伴随细胞内Ca2+超载[49]。
ERS与自噬之间也存在串扰机制[50]。具体而言,UPR在ERS下被激活,以协调的方式与自噬相互作用。UPR和自噬之间的协调可以减轻蛋白质错误折叠及其导致的肾脏损害[40]。在哺乳动物细胞中,PERK-eIF2α-ATF4信号传导途径参与了ERS和自噬串扰的调控[51]。同样有研究发现,可以通过调节动物模型中的PERK,实现ERS和自噬的串扰[52]。
此外,某些物质会通过调节PERK-eIF2α-ATF4信号通路发挥对肾脏的保护作用。研究发现,使用GSK(一种PERK特异性抑制剂),可阻断eIF2α-ATF4信号传导,改善铜诱导的肾小管上皮细胞中的内质网形态损伤,PERK抑制剂可能是治疗肾损伤的良好选择[49]。糖尿病肾病中,在内质网应激条件下,葛根素可通过上调PERK-eIF2α-ATF4信号通路,增强自噬活性,逆转肾小球塌陷和萎缩,改善肾损伤[52]。还有研究发现,白杨素(5,7-二羟基黄酮)可阻断PERK-eIF2α-ATF4-CHOP的上调,减少肾足细胞对ERS产生的UPR,阻断与足细胞凋亡相关的ERS反应,并减少体外和体内裂隙隔膜蛋白的合成,靶向ERS可能是白杨素介导的相关肾足细胞损伤功能改善的潜在机制[53]。以上研究表明,PERK抑制剂可能会对肾病的有效治疗发挥关键作用。
2.3 PKR
PKR是一种普遍表达的丝氨酸/苏氨酸激酶,它的激活与过量摄入营养物质以及多种形式的应激信号(例如机械应激、代谢应激、氧化损伤和细胞因子)有关[7],是炎症和凋亡诱导剂。炎症可能是导致肾纤维化的潜在机制之一[54]。在纤维蛋白生成过程中,肾单位逐渐丧失和组织重塑后,受损的肾小管细胞凋亡,毛细血管渗透,趋化因子或细胞因子等炎性介质与成肌纤维细胞相互作用,招募更多的炎性细胞迁移至肾间质,从而引起进行性肾小球硬化、肾小球膜增生以及肾纤维化[54-55]。PKR参与包括NF-κB和炎症细胞激酶JNK在内的几种细胞途径的调节和维持,整合炎症信号传导,并通过促进不同的促细胞凋亡因子表达,诱导细胞凋亡[56-57]。研究发现,PKR在N-硝基-L-精氨酸甲酯(NG-nitro-L-arginine methyl ester,L-NAME)引起的肾损害和肾小管细胞凋亡的发病机理中起核心作用[58],靶向性PKR治疗可能是治疗高血压相关肾脏并发症的有效方法。
此外,在体内和体外研究中,PKR的激活与多种病理事件的发生有关,例如胰岛素抵抗、葡萄糖耐受不良、糖尿病和心血管并发症中炎症或氧化应激标志物升高[59]。它的激活对胰岛素信号通路、脂肪细胞异常或慢性代谢性炎症产生负面影响。但值得注意的是,PKR的敲除或沉默可显著减弱或抑制上述不良影响[60-61]。研究发现,在高果糖体外培养的大鼠肾上皮细胞中PKR表达显着增加,肾上皮细胞的氧化损伤和细胞凋亡显著性增加,但PKR的选择性抑制剂C16显著减弱了高果糖诱导的对肾细胞的破坏性作用[62]。因此,开发出更安全的PKR抑制剂药物可能是对抗肾脏疾病的新型治疗策略。
2.4 HRI
HRI作为终末红细胞生成的主要调节因子,可被一氧化二氮、渗透胁迫、热休克或缺铁、砷、镉和铅暴露等诱导的外源应激激活,使eIF2α磷酸化,减少三元复合物的形成,抑制mRNA的翻译[21]。HRI的激活可以诱导下游效应因子,包括ATF-4和CHOP,减少氧化应激,促进红细胞生成过程中的红细胞分化作用[20]。研究发现,HRIeIF2α-ATF4通路被激活是红细胞分化所必需的[21,63]。
对于HRI的研究,大多数都集中在红系细胞中,HRI对红细胞分化起保护作用。然而,研究也已证实HRI在巨噬细胞中表达并在巨噬细胞的成熟过程中发挥重要作用[22]。研究发现,在脂多糖处理诱导的应激条件下,激活HRI-eIF2α-CHOP是巨噬细胞成熟过程中所必需的[64]。表明在应激条件下,HRI除影响红细胞外,还可能影响其他谱系的存活和分化。
HRI的激活(以及由此产生的eIF2α磷酸化)可能具有细胞保护作用。有研究发现,在镉处理的小鼠中,肾脏结构显著改变,近端和远端小管破裂以及肾小球塌陷,整个肾脏表现出明显的空泡化、变性和萎缩,其中白细胞浸润是造成肾脏损伤的重要原因之一[65]。然而,HRI缺乏会损害单核细胞和中性粒细胞的发育,减轻肾脏损伤的加重。表明在镉诱导的应激条件下,HRI在白细胞分化和成熟过程中有重要作用,HRI可能参与了抗氧化应激信号传导过程[65]。关于HRI驱动不同类型白细胞分化成熟的具体机制仍需进一步研究,但研究至少表明HRI缺乏导致的分泌细胞因子减少可能是镉治疗后减轻小鼠肾脏损伤的有效途径之一。提示HRI在诱导单核细胞和中性粒细胞保护肾脏免受镉损伤方面发挥重要作用,可能有助于识别预防和治疗镉诱发肾脏疾病。
3 结语与展望
近年来,随着对eIF2α激酶家族(GCN2、HRI、PERK、PKR)的结构功能和作用的深入研究,eIF2α激酶家族参与多种疾病调控的复杂机制越来越受到关注。一方面eIF2α激酶家族的特殊性与多样性,使得它能够与多种分子相互结合并发挥不同的作用。另一方面,eIF2α激酶家族展现出的自身调控的复杂性以及在肾脏中的双面性,因此,eIF2α激酶家族在肾脏疾病中可能发挥重要作用。而肾脏疾病的发生机制十分复杂,是多因素综合作用的结果,有关eIF2α激酶家族直接作用于具体肾脏疾病中的机制研究仍有待深入,尤其是eIF2α激酶家族成员之间的相互代偿性影响仍在研究起步阶段。随着转录组学研究的兴起,RNA免疫共沉淀测序技术、RNA交联免疫共沉淀技术及其相关衍生技术的发展,可更多地探索eIF2α激酶家族相关蛋白质及其对应mRNA与肾病的关系,有助于进一步揭示其参与具体肾脏疾病的分子机制,这将为肾脏疾病的研究提供新思路。未来深入研究eIF2α激酶家族在肾脏疾病中的作用,有望对肾脏疾病的临床诊断和治疗产生积极影响,能使其早日应用于临床,从而造福于人类。
