黄河三角洲贝壳堤岛滨海湿地沉积物细菌群落结构多样性特征
2022-08-17赵凤娟赵自国夏江宝高春明
赵凤娟,赵自国,夏江宝,高春明
(1.滨州学院生物与环境工程学院,山东 滨州 256603;2.山东省黄河三角洲生态环境重点实验室,山东 滨州 256603;3.山东省黄河三角洲野生植物资源开发利用工程技术研究中心,山东 滨州 256603)
贝壳堤是淤泥质或粉砂质海岸特有的一种滩脊类型,在黄河三角洲地区分布广泛.湿地介于水体和陆地之间,兼有水陆特征,被誉为“自然之肾”[1].滨海湿地位于陆地和海洋生态系统的交错过渡地带[2],在保护海岸、防止海水入侵、调节气候和流量、补充地下水、促进元素生物地球化学循环和维护生物多样性等方面发挥着重要作用[3].黄河三角洲贝壳堤岛滨海湿地是具有独特结构与复杂功能的生态系统,已经被划入国家级自然保护区.微生物是湿地生态系统的重要组成部分,作为湿地生态系统的分解者,不仅制约着潮间带湿地类型的分异和演替[4],还在维持湿地物质循环和能量流动方面发挥着不可或缺的作用[5],因而,在湿地沉积物微环境中扮演着极为重要的角色,如去除污染物、分解植物残体以及促进各类营养元素的物质循环等.为揭示湿地生物反应器这个“黑匣子”的运行机制,亟须解析湿地沉积物微生物群落结构特征,为了解湿地环境中微生物与环境因子、微生物与植物、微生物之间的相互关系提供数据支撑.
目前,黄河三角洲滨海湿地研究主要集中在景观格局空间演变或退化[6-7],土壤有机质储量估算[8],水盐胁迫下植被的C、N循环[9],遗传变异及耐受机制[10-12]以及生态地质安全评价和修复[13-14]等方面,而对贝壳砂生境潮间带沉积物微生物群落结构的研究还鲜有报道.贝壳堤岛是由潮间带的死亡贝类壳体及碎屑,通过波浪搬运到高潮带附近堆积而成,贝壳砂土粗砂粒含量高、土壤孔隙度大,在滨海湿地海陆交互、海岸带底质和物质环境呈梯度变化的特殊环境中,可能有特色的微生物群落、生态分布以及功能基因资源[15].因此,本研究应用不依赖培养的PCR-DGGE技术,结合构建16S rDNA文库,分析贝壳堤岛滨海湿地沉积物微生物遗传多样性和系统发育多样性,阐明微生物群落结构多样性.本研究对开发黄河三角洲滨海湿地功能微生物资源和保护该区特色湿地生态系统具有重要意义,也将为针对该区湿地的环境微生物修复技术研发提供参考依据.
1 样线设置与研究方法
1.1 研究区概况
研究区位于黄河三角洲腹地山东省滨州市无棣县西北部的典型贝壳堤岛——棘家堡子岛,地理坐标为38°13′6″~38°14′8″N,117°56′5″~117°57′14″E.贝壳堤岛的整体海拔多在5 m以下;土壤类型以滨海盐土类和贝壳砂土为主,主要由风积物和钙质贝壳土壤化成土;地下水矿化度较高,埋深浅;该区气候四季分明,属暖温带大陆性季风气候,雨热同期,年降水量为550 mm,年蒸发量为2 430.6 mm,年平均气温为12.36 ℃[16];植物群落主要是盐生、旱生灌草丛或少量的水生草本群落.
1.2 样线设置及样品采集
采样地点为贝壳堤岛的典型滨海湿地区域,沿自陆向海的方向设置4个样线(0,1,3,5),见图1,每个样线在高潮带、中潮带和低潮带分设3个采样点,共计12个采样点.取样深度0~10 cm,每个样点取3次,将其混合为一个样品,编号.具体的采样点坐标和样品编号见表1.采样时使用无菌铁铲,将沉积物样品铲入事先处理的无菌密封塑料袋或EP管中,迅速放入便携式冰盒中保存.将样品带回实验室,过筛(孔径2 mm),除去可见动物残体、贝壳或砂砾等,过筛后的沉积物多数自然风干后,进行理化性质测定;另外各取5 g保存到-40 ℃条件下,用于后续沉积物微生物群落结构研究.
1.3 沉积物理化指标测定
沉积物样品的5个理化指标为pH、盐度、有机质、总氮(TN)、总磷(TP),测试结果见表2.
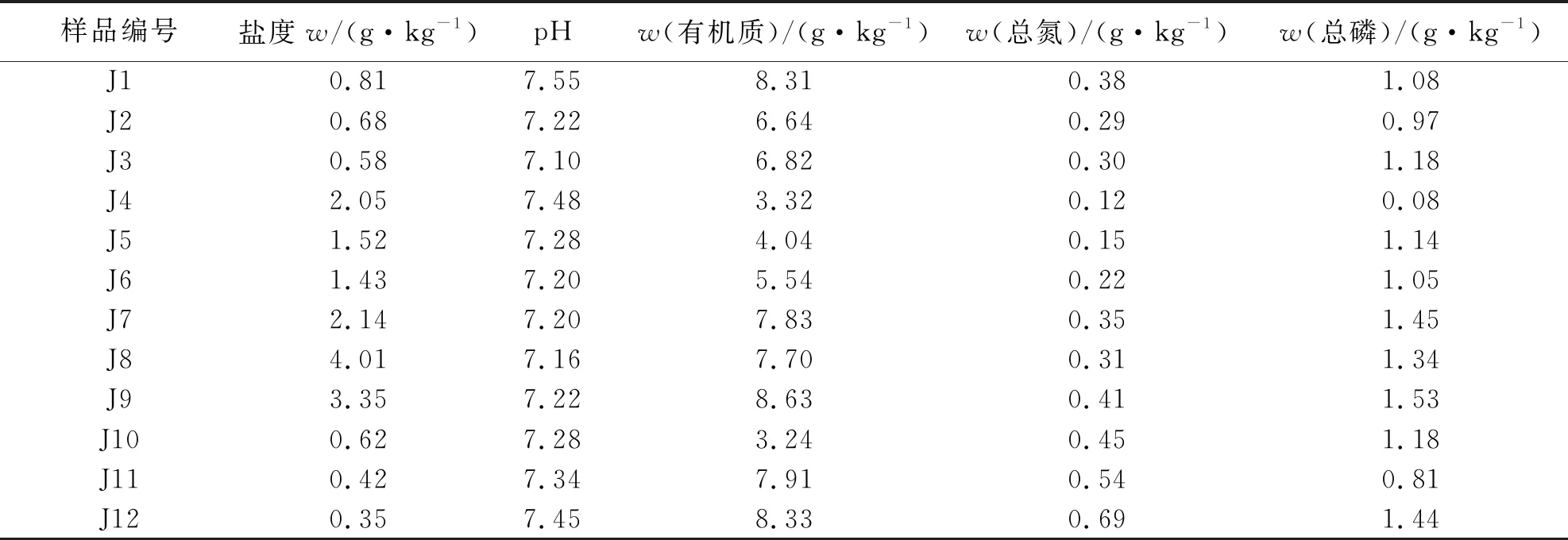
表2 供试沉积物样品理化指标
1.4 沉积物微生物总DNA提取
DNA提取利用Fast DNA Spin Kit for Soil 试剂盒(50)(MP公司,美国)完成.量取解冻后的1 mL沉积物样品,用PBS缓冲液清洗离心(14 000 g)两次,按照试剂盒产品说明书提取总DNA,保存于-20 ℃.通过0.8%琼脂糖凝胶电泳检测获得总DNA样品质量,通过Nano Drop ND-1000 微型分光光度计测定总DNA浓度.
1.5 16S rDNA V3区片段PCR扩增
将所得高质量基因组DNA作为聚合酶链式反应(PCR)模板,选用对大多数细菌的16S rRNA 基因V3区具有特异性的引物[16]GC-341F和534R进行PCR扩增,目的扩增产物片段长度约194 bp.总细菌PCR引物信息见表3.

表3 总细菌PCR引物信息
所用50 μL PCR 反应体系组成:37.75 μL 灭菌高纯水,5 μL 10×buffer,4 μL dNTP混合物(各2.5 mmol/L),正、反向引物(10 pmol/μL)各1 μL,0.25 μL的Taq DNA聚合酶(5 U/μL,TaKaRa),模板DNA 1 μL.
采用配对PCR策略,第1次PCR为梯降PCR,扩增反应条件为94 ℃预变性5 min,94 ℃变性1 min,65 ℃退火1 min,72 ℃复性2 min,共20个循环,每个循环退火温度下降0.5 ℃;94 ℃变性1 min,55 ℃退火1 min,72 ℃复性2 min,5个循环,共25个循环;72 ℃延伸10 min.第1次PCR产物稀释10倍,作为第2次PCR扩增模板,进行第2次PCR,扩增条件为94 ℃预变性5 min,94 ℃变性1 min,55 ℃退火1 min,72 ℃复性2 min,共3个循环;72 ℃延伸6 min.PCR 产物用1.5%琼脂糖凝胶电泳检测,用于后续DGGE分析.
1.6 PCR反应产物变性梯度凝胶电泳(DGGE)分析
使用浓度为8%的丙烯酰胺胶(PAGE)对总细菌PCR扩增片段进行电泳,设置变性梯度范围为35%~55%.所用DGGE仪为Bio-Rad DGene system(Bio-Rad,美国),所用电泳缓冲液为1×TAE,运行参数设置为60 ℃,50 V,运行10 min;130 V运行7 h.电泳结束后,用1∶10 000的Gelred核酸染料(Biotium公司,美国)染色30 min,去离子水漂洗2次;在凝胶成像系统(Molecular Imager Gel DOCTM XR System,Bio-Rad,USA)紫外光(470 nm)照射下观察并照相.用Quantity One 4.3.6 软件(Bio-Rad,USA)对所得DGGE 图谱进行统计分析.
选择香浓-维纳多样性指数(Shannon-Wiener diversity index,H′)[17]比较各样品的细菌多样性:H′=-∑PilnPi,式中:Pi=ni/N,ni为第i条条带的多度,N为样品中所有条带的总多度.
采用Quantity one 软件中的非加权组算术平均法(UPGMA,unweighted pair-group method with arithmetic means)分析比较不同沉积物样品微生物群落结构的相似性.
1.7 DGGE条带序列测定及系统发育树构建
目标DGGE条带回收后,先在紫外灯下将目标条带整体切割,对应放入编号的EP 管中,用去离子水冲洗两次,用50 μL TE buffer浸泡,放置于4 ℃过夜,使DNA溶解于TE buffer中.取回收后的DNA作为PCR模板,用不带GC夹的DGGE引物(341F和534R)进行特异性PCR扩增,扩增条件和体系同第1次PCR,然后送往诺赛基因(北京)公司进行测序.所用载体为pMD19-T载体,转化细胞为 Trans5α化学感受态细胞.若样品测序结果呈现非特异性,则将非特异性条带进行克隆测序,以便进一步分析其群落结构.TA克隆由诺赛基因(北京)公司参照pMDTM19-T Vector Cloning Kit说明书进行.
将所得序列以Blast 程序在GenBank中进行相似性比较分析,选择与数据库中16S rDNA序列同源性最高的序列作为参照菌株.
将所有序列用标准模式导入,使用Clustalx 1.83软件进行序列对比,发现序列方向差异;使用EditSeq软件将所有序列方向调整一致;使用Editplus 3进行建树前的建模和模型计算分析;使用Mrbayes进行系统进化树构建[18];使用treeview软件查看构建的进化树.
1.8 微生物多样性与环境因子相关性分析
运用统计学软件PSW Statistics 18.0对DGGE图谱量化后的数据和环境理化指标数据进行主成分分析(PCA,Principle Component Analysis),通过相关性分析,检测不同样线沉积物样品微生物多样性与其环境因子的相关性.
2 结果与分析
2.1 沉积物样品总DNA提取及V3 区扩增
通过FastDNA Spin Kit for Soil 试剂盒提取获得的沉积物基因组DNA均在23 kb处有明显的主带,且无弥散或拖尾现象;以引物对(GC-341F和534R)进行的V3区PCR扩增均能够扩增出大小约为200 bp的DNA片段,且无明显非特异性扩增条带.
2.2 沉积物样品细菌群落遗传多样性
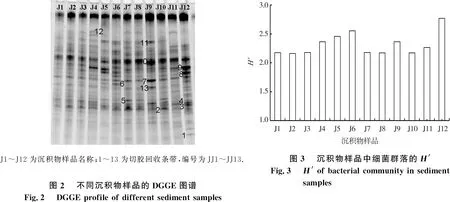
对所有沉积物样品16S rDNA的V3区进行扩增,并对扩增产物进行变性梯度凝胶电泳检测,所得DGGE指纹图谱见图2.DGGE图谱中,电泳条带的数量可以直观地反映出样品中细菌群落的遗传多样性.其中,不同细菌16S rDNA V3区基因片段通过不同条带显示,条带亮度反映出每个泳道中细菌相对生物量的大小.
利用Quantity One软件分析电泳图谱中每条条带信息,对各样品中的细菌多样性指数进行统计分析,结果见图3.由图3可见:0号样线多样性指数相对较低,且高、中、低潮带样品多样性指数差异不大,但在1、3、5号样线都发现位于低潮带的沉积物样品,其H′较高.
2.3 沉积物样品细菌群落相似性
不同沉积物样品之间细菌群落相似性比较是通过Quantity One软件统计分析DGGE指纹图谱中不同样品之间共有条带数目实现的.沉积物样品的细菌群落结构相似性聚类分析结果见图4.由图4可知:从总体聚类来看,贝壳堤岛滨海湿地沉积物微生物群落结构多样性较为丰富,12个沉积物样品聚为两大簇,5个类群;位于同一样线的样品倾向于越早聚类在一起,比如,J1、J2和J3(0号样线)聚类在同一个类群;J7和J8首先聚类成一个小分支,然后很快与J9聚类在一起;J10、J11和J12(5号样线)位于临近的类群等.
2.4 沉积物样品回收条带序列的系统发育分析
对沉积物样品J1~J12的PCR-DGGE 条带进行比较筛选,13条优势条带通过切胶回收(图2),对其进行序列测定.其中,JJ-7、JJ-9呈现较好的特异性,而其余11条测序呈现非特异性结果,通过Ta克隆进一步深入检测群落结构多样性.克隆测序结果显示,每条条带可获得13~35个阳性克隆,共计215条不同序列.每条非特异性条带随机选取两条克隆测序结果,例如,JJ-1选择JJ-1-8和JJ-1-14,加上JJ-7和JJ-9,共计24条序列,合并进行系统发育树构建,见图5.

利用Blast 程序对所得序列在GenBank中进行相似性比较分析.结果表明,22条序列与数据库中16S rDNA序列的相似性可达97%~100%,只有2条相似性稍低,为94%,并且发现,同源性最高的序列多数为未培养微生物(表4).选择同源性最高的序列作为鉴定参照菌株,结果显示,来自同一条带的Ta克隆序列的系统发育关系可以相距很远,如JJ-3-3和JJ-3-20;而来自不同条带的序列,可能在系统发育树上关系很近,如JJ-7和JJ-3-3.从鉴定水平来说,有些只能鉴别到门,如Bacteroidetes(拟杆菌门)、Sphingobacteria(鞘脂杆菌门)、Chloroflexi(绿弯菌门),多数能鉴别到属,分属proteobacterium(变形菌属)、Salegentibacter(需盐杆菌属)、Pseudomonas(假单胞菌属)、Marinobacter(海杆菌属)、Halomonas(盐单胞菌属)、Sulfitobacter(亚硫酸杆菌属)、Cytophaga(噬细胞菌属)、Flavobacterium(黄杆菌属)、Fusobacteria(梭菌属)等,还有一类属于Actinobacteridae(放线菌亚纲).有的(如JJ-11-15)同为DGGE电泳所得序列,为未知种属未培养的细菌序列.

表4 部分测序序列BLAST分析
2.5 环境因子与样品微生物群落多样性关系
通过主成分分析提取到两个主成分:PC1(39.2%)和PC2(27.0%),累计贡献率为66.2%.以PC1和PC2分别为横轴和纵轴,以4个样线为分组,将12个样品做成散点图,见图6.由图6可见:来自不同样线的沉积物样品可以很好地区分开.但相关性分析发现,单个环境因子与遗传多样性之间的相关性不强,相关系数均小于0.5.

图6 环境变量与DGGE揭示的微生物群落多样性PCA分析
3 讨 论
本研究结果显示:滨海湿地沉积物微生物多样性香浓-维纳指数以5号样线低潮带样品最高;除0号样线高、中、低潮带的差异不大外,在样线1、3、5都有发现,其低潮带样品的多样性指数更高,说明低潮带的微生物群落相对更为丰富.分析原因,可能与贝壳堤岛潮间带底质由贝砂质、泥沙质逐渐过渡至淤泥质有关,更有利于有机质积累,单个样线沉积物在潮间带都呈现出有机质含量自陆向海递增的趋势也与此有关.而有机碳、氮含量被认为是微生物生长的主要限制因素[19-20].
根据DGGE指纹图谱,各沉积物样品细菌群落结构相似性的UPGMA聚类结果可以反映出位于同一样线的样品倾向于更早聚类在一起.耦合沉积物细菌多样性与沉积物理化指标,通过主成分分析提取到两个主成分:PC1(39.2%)和PC2(27.0%),累计贡献可以解释各样线沉积物样品66.2%的差异,在按照主成分为横、纵坐标所做的PCA散点图中,可以明显区分不同样线的沉积物样品分布,反映出与UPGMA聚类图相同的趋势.而多重相关性分析显示,在多样性指数和单个理化指标参数之间未检测到显著的正或负相关关系,这可能与各采样地点,尤其是不同样线之间的环境指标差异较大有关.
DGGE指纹图谱之所以成为一个研究细菌群落结构变化趋势的常用方法,是因为它能够直观地反映出不同沉积物样品之间细菌组成成分的差异,借助对DGGE条带进行切胶回收测序便可实现对细菌群落结构的多样性组成检测[21].本研究对不同样线的沉积物样品DGGE切胶回收的优势条带进行测序,其中,JJ-7和JJ-9两条回收条带测序获得了特异性结果,显示其分别属γ-变性菌属和黄杆菌属.其中,γ-变性菌属属于变形菌门,该类群具有很强的适应性,具有解磷作用;而黄杆菌属是以产生黄色素为特征的一个菌属,为条件致病菌,可引起肺炎、脑膜炎或败血症等,它们在淡水、海水和土壤(沉积物)等不同环境中均有分布[22].其余11条回收条带测序呈现非特异性扩增结果,因而通过Ta克隆进一步深入检测其群落结构多样性,克隆测序结果显示,每条条带可获得13~35个阳性克隆,共计215条不同序列.尽管从技术分析精度和深度角度来看,对于微生物多样性较高的环境样品,PCR-DGGE技术与新兴的各类高通量测序技术平台相比,在检测的微生物种类,以及门、属水平的热度分布等方面不具优势,但其成本低、用时少,通过耦合切胶回收及后续Ta克隆仍然可以实现对来自不同样品的微生物群落多样性进行直观比较,并可对其微生物的优势种类实现有效分类和甄别[23].
通过在GenBank中进行BLAST比较分析可知,所有同源序列均得到了较高的相似性,多数在97%~100%,最低的同源性为94%.分析其系统发育关系分布可知,多数属于变形菌门,α、β和γ都有分布,属于解磷菌,这与吴文卫等[23]、吴玲[24]的研究结论一致;也有属于鞘脂杆菌门,可以降解维生素,在海洋细菌中占有明显优势;绿弯菌门,是一类可以通过光合作用产生能量的细菌;而海杆菌属中的一些种类被报道为石油降解菌[25];还有一部分属于病原菌,例如拟杆菌门,其中有些种类常见于各类粪便;假单胞菌属的一些杆菌,抗药性很强,能引起伤口化脓性病变.近年来的海岸带开发,如人工养殖及生态旅游项目等,也给潮间带微生物群落构成带来了一定影响.总体来看,贝壳堤岛滨海湿地沉积物中功能微生物资源丰富,且通过自身代谢功能以及群落之间的协同或拮抗作用,经长期适应滨海湿地的特殊环境,才能成为优势菌种.
经鉴别的微生物绝大多数为未培养微生物,要具体了解这些优势菌群在原位生态环境中的生理生化特性及其在贝壳堤滨海湿地生态系统中的结构与功能,还需要进行更加全面细致的试验设计,利用精度更高的检测技术或平台,如Illumina等高通量测序手段[25],或开发新型微生物培养技术,并可借助BIOLOG微平板技术等[26],对微生物群落功能开展深入研究.本研究借助PCR-DGGE技术,初步明晰了黄河三角洲贝壳堤岛滨海湿地沉积物微生物群落多样性及其结构,发现该区存在大量未培养和尚未鉴定出的微生物菌种资源,这对于贝壳堤岛特色微生物资源的开发和黄河三角洲滨海湿地生态系统保护与修复都具有重要意义.
4 结 论
1)黄河三角洲贝壳堤岛滨海湿地沉积物微生物多样性指数在4个样线上都以低潮带微生物群落多样性更高,与潮间带底质由贝砂质、泥沙质逐渐过渡至淤泥质有关;沉积物样品细菌群落结构相似性UPGMA聚类分析结果与耦合沉积物细菌多样性和沉积物理化指标所做PCA主成分分析结果趋势相同,即位于同一样线的样品倾向于聚类在一起.
2)通过PCR-DGGE耦合切胶回收及后续Ta克隆测序对不同沉积物样品微生物的优势种类实现了有效分类和鉴别,成功构建了系统发育树;借助GenBank中BLAST比较,多数能够鉴定到属,优势种类来自变形菌门、鞘脂杆菌门、绿弯菌门和海杆菌属等与解磷、降解纤维素、光合产能或石油烃类降解相关的菌属;也有一定的病原菌分布,如拟杆菌门和假单胞菌属的一些种类.
3)本研究借助PCR-DGGE技术,初步明晰了黄河三角洲贝壳堤岛滨海湿地沉积物微生物群落多样性及其结构,对于贝壳堤岛特色微生物资源的开发和滨海湿地生态系统的保护与修复都具有重要意义.经鉴别的微生物绝大多数为未培养微生物,需借助精度更高的检测技术或平台,开发新型培养技术,开展进一步深入研究.
