新型脯氨醇N-氧化物类HDAC抑制剂的设计合成*
2022-08-16支泰雷李勇军
陈 蕾,支泰雷,李 燕,李勇军,何 彬
(1 贵州省药物制剂重点实验室,贵州 贵阳 550004;2 贵州医科大学基础医学院,贵州 贵阳 550004;3 民族药与中药开发应用教育部工程研究中心,贵州 贵阳 550004)
肿瘤是一类严重威胁人类健康与生命的重大疾病。由于肿瘤的发病机理错综复杂,针对肿瘤的药物作用靶点很多。近年来,组蛋白去乙酰化酶(HDAC)成为抗肿瘤药物作用的重要靶点。HDACs是一类广泛存在于真核细胞中的蛋白酶,参与染色体的结构修饰和对其他功能蛋白的条件[1-2],在基因表达[3]、细胞生长[4]、分化和凋亡[5]中发挥着重要的作用。因此,以HDACs为靶点设计小分子抑制剂被视为治疗肿瘤的一个有效策略。HDAC抑制剂已成为一种具有良好前景的抗肿瘤药物。2006年12月,FDA批准SAHA(Suberoylanilide hydroxamic acid, Vorinostat)上市。这是第一个上市的HDAC抑制剂,用于治疗皮肤T淋巴细胞淋巴瘤。西达本胺(Chidamide,商品名爱谱沙/epidaza)属于全新作用机制的综合靶向抗肿瘤靶向药物,于2015年1月获准全球首个获准上市的亚型选择性组蛋白去乙酰化酶口服抑制剂,广泛用于临床治疗。
根据他们与酵母HDACs的同源性、细胞内定位和组织分布特异性又可分为四种类型:Class I,Class Ⅱ,Class Ⅲ,ClassⅣ,如图1所示。其中Class I,Class Ⅱ,ClassⅣ是最基本的组蛋白去乙酰化酶,共包括11个亚型。Class I 包括HDAC1,HDAC2,HDAC3,HDAC8亚型;Class Ⅱ包括HDAC4-7,9-10亚型;ClassⅣ包括HDAC11亚型。Class Ⅲ是基于酵母Sirt2的类似蛋白。Class I,Class Ⅱ,ClassⅣ三类是Zn2+的酶,在其底部有一个含Zn2+的催化口袋,可以结合锌离子螯合基团(ZBG)[6-8]。

图1 HDACs家族[8]Fig.1 HDAC family
HDACs抑制剂能够抑制HDACs活性,增加组蛋白乙酰化程度,提高抑癌基因表达水平,因此,能够抑制肿瘤细胞的增殖、诱导细胞分化和凋亡[9-10]。X-射线晶体衍射法结构分析HDACIs具有三个功能区域:(1)锌离子结合区(zinc binding group,ZBG)与酶的活性位点的Zn2+,并与活性中心氨基酸作用;(2)连接器(linker)占据活性位点的狭长的通道,它的链长直接影响到锌离子结合区与的结合情况;(3)表面识别区(cap group)与酶活性口袋边缘的氨基酸残基作用,决定抑制剂分子对酶的识别及结合程度,辅助锌离子结合区与Zn2+螯合[8,11]。
作为第一个被美国食品药物管理局(FDA)批准上市的HDACI,化合物SAHA(suberanilohydroxamic acid)具有较高的酶抑制活性,对Ⅰ类和Ⅱ类HDACs均具有很好的抑制效果。另一个上市的药物FK228,其二硫键在体内水解为巯基,作为抑制剂的锌离子结合区与酶活性中心Zn2+和氨基酸作用[11],如图2所示。
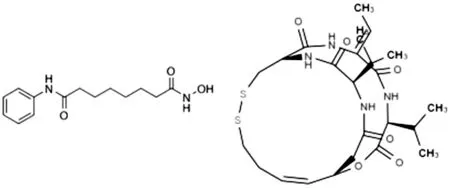
图2 SAHA和FK228的结构Fig.2 Structure of SAHA and FK228
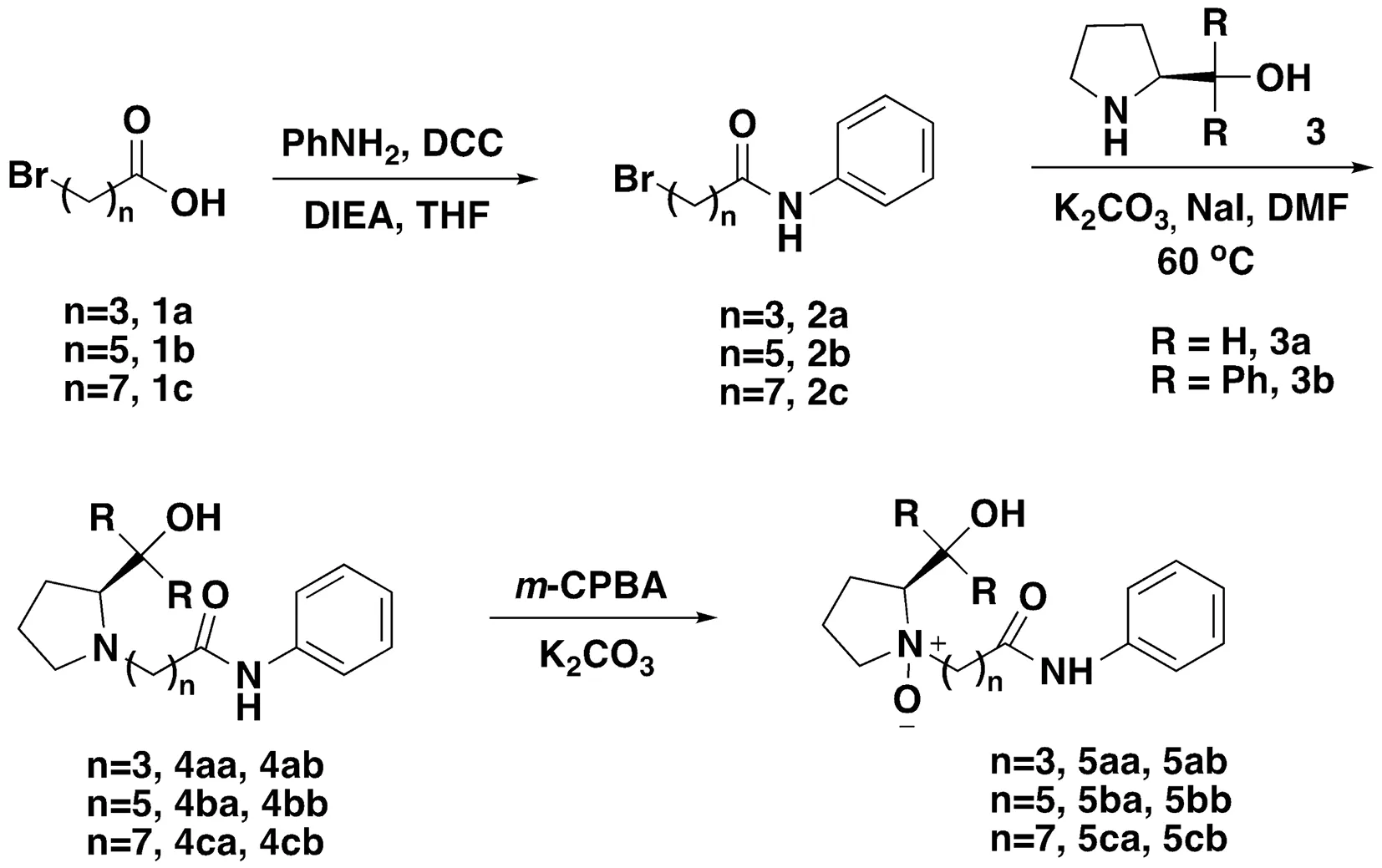
图3 新型脯氨酸抑制剂5的合成路线Fig.3 Synthetic route of new proline inhibitor 5
分别以4-溴丁酸,6-溴己酸,8-溴辛酸为原料,设计合成一系列的可能具有良好活性的N-氧化物类HDAC抑制剂,探索ZBG和Linker的变化导致的活性变化,如图3所示。并可从中寻找活性较好的药物作为先导化合物进行结构修饰和改造,为后续合成打好铺垫。
1 仪器与试剂
1.1 仪 器
WGLL-125BE 鼓风干燥箱,天津市泰斯特仪器有限责任公司;GB3002 电子天平,METTLER TOLEDO;EYELA N-1100 旋转蒸发仪,上海爱朗仪器有限公司;JNM-ECS400 核磁共振测试仪,JEOL公司;UHPLC-MicroTOF-QⅡ 高分辨质谱仪,Bruker Daltonics公司;XODC-0506 低温恒温槽,南京先欧仪器制造有限公司;B11-1 控温磁力搅拌器,上海司乐仪器有限公司;2XZ-1 智能真空泵,浙江黄岩黎明有限公司。其他:茄形瓶,恒压滴液漏斗,分液漏斗,水浴锅,烧杯,量筒,滴管,结晶滤头,移液管等。
1.2 试 剂
4-溴丁酸(98%)、6-溴己酸(98%)、8-溴辛酸(98%)、苯胺(99%)、碘化钠(99%),阿拉丁试剂;二氯亚砜、DCC(二环己基碳二亚胺)、DIEA(N,N-二异丙基乙胺)、L-脯氨醇、(S)-(-)-α,α-二苯基脯氨醇、K2CO3、DMF(N,N-二甲基甲酰胺),广东光华科技股份有限公司;THF(四氢呋喃)、m-MCPA(间氯过氧苯甲酸)、柱层析硅胶(200-300目,100-200目)、薄层层析硅胶GF254、实验所用试剂或溶剂在反应前均按反应要求进行预处理,所有起始原料均为市售化学纯或试剂纯。
2 方法与结果
2.1 化合物2的制备
在100 mL的茄形瓶加入1.0 g的4-溴丁酸1a,适量THF作为反应溶剂,冰浴下滴加1.2 eq的二氯亚砜,常温下反应 2 h后,再加入1.2 eq的苯胺,约2 h后监测反应完全,将反应体系中的溶剂减压蒸馏除去,用二氯甲烷萃取,饱和食盐水洗多次,无水硫酸钠干燥。得到油状混合物2.5 g。采用石油醚:乙酸乙酯=10:1极性的洗脱剂进行洗脱。得到2a为720 mg。收率为50%。1H-NMR(400 MHz, CDCl3)δ(ppm)7.50(d, 2H); 7.29(m, 2H); 7.117(t, 1H); 3.64(m, 2H); 2.49(m, 2H); 1.19(m, 2H);13C-NMR(100 MHz, CDCl3)δ 129.04, 124.40, 119.73, 50.90, 44.48, 34.09, 27.83。
取100 mL的茄形瓶,加入1.0 g 6-溴己酸1b,1.2 eq的苯胺,以及1.2 eq的DCC和1.2 eq的N,N-二异丙基乙胺(DIEA),加入5 mL 的四氢呋喃,常温下反应5 h,TLC监测反应完全。将反应体系中溶剂减压蒸尽,采用500 mL二氯甲烷萃取,100 mL饱和食盐水洗4次。收集有机层,采用无水Na2SO4干燥。采用柱层析石油醚:乙酸乙酯=15:1的洗脱剂分离得到1.4 g浅黄白色固体的化合物2b,收率为86%。1H-NMR(400 MHz, CDCl3)δ(ppm)7.48(d, 2H); 7.31(d, 2H); 7.09(m, 1H); 3.409(t, 2H); 2.38(t, 2H); 1.87(m, 2H); 1.75(m, 2H); 1.54(m, 2H);13C-NMR(100 MHz, CDCl3)δ 129.02, 124.26, 119.70, 37.45, 33.65, 32.42, 27.70, 24.59。
取100 mL的茄形瓶,加入1.0 g 8-溴辛酸1c,1.2 eq的苯胺,以及1.2 eq的DCC和1.2 eq的DIEA,采用THF作为反应溶剂,常温下反应5 h,TLC监测反应完全。将反应体系中溶剂减压蒸尽,采用500 mL二氯甲烷萃取,饱和食盐水洗 (100 mL×4)。收集有机层,采用无水Na2SO4干燥,采用石油醚:EtOAc=10:1的洗脱剂洗脱,得到化合物2c为0.75 g。收率为56%。1H-NMR(400 MHz, CDCl3)δ(ppm)7.49(d, 2H); 7.28(d, 2H); 7.10(m, 1H); 3.37(m, 2H); 2.34(m, 2H); 1.87(m, 2H); 1.70(m, 2H); 1.35(m, 5H);13C-NMR(100 MHz, CDCl3)130.09, 124.20, 119.67, 50.91, 37.72, 33.98, 32.65, 29.01, 28.49, 27.94, 25.41。
2.2 化合物4的制备
在化合物2a中加入1.2 eq的L-脯氨醇3a或3b,2.0 eq的K2CO3以及1.0 eq的NaI,采用DMF作为溶剂,50~70 ℃条件下反应,约6 h后TLC监测反应完全。将反应体系中溶剂减压蒸馏,采用乙酸乙酯萃取,饱和NaCl水溶液多次洗,采用无水Na2SO4干燥,得到油状混合物,采用二氯甲烷:甲醇=10:1得到化合物4aa为108 mg。收率为50%。1H-NMR(400 MHz, CDCl3)δ(ppm)7.64(m, 2H); 7.24(m, 2H); 7.07(m, 1H); 3.99(m, 1H); 3.90(m, 2H); 3.67(m, 2H); 3.49(m, 2H); 3.14(m, 1H); 2.81(m, 2H); 1.91(m, 2H); 1.23(s, 2H);13C-NMR(100 MHz, CDCl3)171.02, 138.5, 128.90, 124.55, 120.22, 70.54, 59.92, 56.67, 55.00, 34.25, 26.38, 23.45, 21.40;HRMS(ESI)m/z: Calculated for C15H22N2O2[M+H]+263.2651, found 262.2681。 采用二氯甲烷:甲醇=20:1洗脱,得到化合物4ab为171 mg。收率为41%。1H-NMR(400 MHz, CDCl3)δ(ppm)7.62(d, 1H); 7.50(m, 2H); 7.27(m, 8H); 7.20(s, 1H); 7.09(m, 2H); 4.41(m, 1H); 3.68(s, 1H); 2.38(m, 1H); 2.08(m, 2H); 1.96(dd, 2H); 1.86(m, 2H); 1.24(s, 3H); 0.85(m, 1H);13C-NMR(100 MHz, CDCl3)171.70, 138.41, 129.32, 129.27, 129.20, 126.37, 56.32, 33.27, 29.17, 26.40, 24.30; HRMS(ESI)m/z: Calculated for C27H30N2O2[M+H]+415.2382, found 414.2307。
取50 mL的茄形瓶 在200 mg化合物2b中加入1.2 eq的L-脯氨醇(3a或3b),2.0 eq的K2CO3以及1.0 eq的NaI,采用DMF作为溶剂,60 ℃条件下反应,约6 h后TLC监测反应完全。将反应体系中溶剂减压蒸馏,水洗多次,乙酸乙酯萃取,用无水Na2SO4干燥,柱层析分离纯化,使用二氯甲烷:甲醇=(30:1)~(15:1)的洗脱剂进行梯度洗脱,分离得到化合物4ba为201 mg,收率93%。分离得到化合物4bb为270 mg,收率82%。化合物4ba:HRMS(ESI)m/z: Calculated for C17H26N2O2[M+H]+290.1994, found 291.1954。化合物4bb:235 mg。收率为75%。HRMS(ESI)m/z: Calculated for C29H34N2O2[M+H]+442.2620, found 443.2529。
取50 mL的茄形瓶 在200 mg化合物2c中加入1.2 eq的 L-脯氨醇(3a或3b),2.0 eq的K2CO3以及1.0 eq的NaI,采用DMF作为溶剂,60 ℃条件下反应,约6 h后TLC监测反应完全。将反应体系中溶剂减压蒸馏,水洗多次,乙酸乙酯萃取,用无水Na2SO4干燥,柱层析分离纯化,使用二氯甲烷:甲醇=(40:1)~(25:1)的洗脱剂进行梯度洗脱,分离得到化合物4ca为181 mg,收率85%。分离得到化合物4cb为235 mg,收率75%。化合物4ca:1H-NMR(400 MHz, CDCl3)δ(ppm)7.97(s, 1H); 7.59(d, 2H); 7.240(d, 2H); 7.04(m, 1H); 3.93(m, 2H); 3.85(s, 1H); 3.50(s, 1H); 3.31(s, 1H); 2.86(m, 2H); 2.37(t, 2H); 2.123(m, 3H); 1.70(t, 3H); 1.36(s, 6H);13C-NMR(100 MHz, CDCl3)171.89, 139.22, 128.85, 123.97, 119.79, 60.15, 54.61, 37.47, 28.21, 27.92, 26.38, 25.97, 25.08, 24.99, 23.67; HRMS(ESI)m/z: Calculated for C19H30N2O2[M+H]+318.2307, found 319.2358。 化合物4cb:235 mg。收率为75%。1H-NMR(400 MHz, CDCl3)δ(ppm):7.61(m, 6H); 7.30(m, 8H); 7.19(m, 2H); 3.2(s, 1H); 2.3(s, 4H); 1.9(s, 4H); 1.7(s, 7H); 1.12(m, 4H);13C-NMR(300 MHz, CDCl3):171.89, 139.22, 128.94, 128.09, 126.38, 125.65, 125.47, 124.04, 119.68, 37.660, 33.913, 29.20, 26.36, 25.56, 24.91 ,24.26; HRMS(ESI)m/z: Calculated for C31H38N2O2[M+H]+471.2921, found 470.2933。
2.3 化合物5的制备
将50 mg的化合物(4aa、4ab、4ba、4bb、4ca、4cb)置于 25 mL的茄形瓶中,加入1.0 eq 的m-CPBA(间氯过氧苯甲酸),2.0 eq 的K2CO3。采用THF作为反应溶剂,冰浴下反应约10 min后,TLC监测反应完全,将反应体系中溶剂减压蒸馏完毕后,采用二氯甲烷萃取,饱和NaCl溶液水洗,无水Na2SO4干燥。采用二氯甲烷:甲醇=20:1的极性的洗脱剂进行洗脱,浓缩得到化合物5aa为24 mg,收率46%。化合物5ba:1H-NMR(400 MHz, CDCl3)δ(ppm)7.90(s, 1H); 7.56(d, 2H); 7.30(m, 3H);7.04(s, 1H); 4.25(m, 2H); 3.89(d, 1H);3.73(m, 3H);2.26(m, 3H);2.03(m, 4H);1.83(d, 2H);1.63(t, 2H); 1.22(d, 7H);13C-NMR(100 MHz, CDCl3): 170.20, 138.17, 128.11, 128.03, 121.60, 71.34, 61.17, 38.40, 26.17, 25.32, 23.12, 19.44; HRMS(ESI)m/z: Calculated for C19H30N2O3[M+H]+334.2256, found 335.2227。
二氯甲烷:甲醇=40:1,浓缩得到化合物5bb为23 mg,收率为44%。化合物5b:1H-NMR(400 MHz, CDCl3)δ(ppm)7.61(m, 6H); 7.30(m, 8H); 7.19(m, 2H); 3.2(s, 1H); 2.3(s, 4H); 1.9(s, 4H); 1.7(s, 7H); 1.12(m, 4H);13C-NMR(100 MHz, CDCl3): 171.85, 130.28, 129.15, 128.42, 127.03, 124.93, 119.96, 77.99, 77.55, 77.07, 68.76, 38.30, 28.58, 28.19, 26.33, 25.43, 20.25; HRMS(ESI)m/z: Calculated for C31H38N2O3[M+H]+486.2882, found 487.2944。
二氯甲烷:甲醇=20:1的极性的洗脱剂进行洗脱,浓缩得到化合物5ca为24 mg,收率为46%。HRMS(ESI)m/z: Calculated for C19H30N2O3[M+H]+335.2227, 334.2256。
二氯甲烷:甲醇=40:1洗脱,浓缩得到5cb为23 mg。收率为44%。1H-NMR(400 MHz, CDCl3)δ(ppm)7.61(m, 6H); 7.30(m, 8H); 7.19(m, 2H); 3.2(s, 1H); 2.3(s, 4H); 1.9(s, 4H); 1.7(s, 7H); 1.12(m, 4H);13C-NMR(100 MHz, CDCl3): 171.85, 130.28, 129.15, 128.42, 127.03, 124.93, 119.96, 77.99, 77.55, 77.07, 68.76, 38.30, 28.58, 28.19, 26.33, 25.43, 20.25; HRMS(ESI)m/z: Calculated for C31H38N2O3[M+H]+487.2944, found 486.2882。
3 结 论
通过考察不同linker链长的HDAC抑制剂,首先以4-溴丁酸,6-溴己酸,8-溴辛酸为起始原料,采用几步简单的酰胺缩合反应、取代反应和过氧化反应后即可制备得到R基团分别由氢原子和苯基两种不同取代的新型N-氧化物脯氨醇类HDAC抑制剂,目标化合物经过核磁共振和高分辨质谱(1H-NMR、13C-NMR、 HRMS)的图谱分析确证。该合成方法简单、操作性强、原料易得,期望进行活性测试后来筛选得到活性更好的目标产物,为后续发展新型的HDAC抑制剂奠定基础。
