基于CRISPR/Cas9 技术构建Plcz1 基因敲除小鼠模型
2022-08-16曹彬彬蔡瑶王宝珠饶喻王超苟克勉王
曹彬彬蔡 瑶王宝珠饶 喻王 超苟克勉王 涛∗
(1.扬州大学兽医学院,江苏 扬州 225009;2.江苏高校动物重要疫病与人兽共患病防控协同创新中心,江苏 扬州 225009)
生殖能力下降是人和动物共同面临的健康问题,不仅对畜牧业造成经济损失,而且对人类健康构成严重威胁。 因此,寻找动物生殖过程中关键的调控基因,探讨其生殖发生过程中的调控机制成为该领域内关注的焦点之一。 在过去的半个多世纪里,许多在动物生殖过程中发挥重要作用的精子因子相继被发现,在动物发生受精时,精子中的某些因子会通过融合孔进入卵母细胞胞质中,启动卵母细胞内钙离子释放系统,从而激活卵母细胞[1]。 研究发现,哺乳动物受精的特征是在精子-卵子融合后,卵子内持续2~4 h 不同程度的Ca2+振荡[2],该过程是恢复第一次减数分裂和胚胎发育的前提,同时还阻止多精入卵的发生[3]。
磷脂酶C zeta(PLCζ)是Saunders 等[4]在研究小鼠睾丸中PLC 时发现的新亚型,随后证实PLCζ不仅存在于睾丸中,并且拥有其他亚型中的基本结构域。 该蛋白结构域由4 个位于N 端的EF 手型结构域、位于中心的特征性X 和Y 催化域以及碳基端的C2 结构域构成[5]。 其中EF 手型区域对低浓度Ca2+敏感,使PLCζ 在静息状态的Ca2+水平具有活性。 通过C2 结构域与PI(3)P、PI(5)P 或一种未知的膜蛋白相互作用可能实现PLCζ 与特定囊泡膜的结合[6],带正电的XY 连接区与第一个EF 手型结构域相互作用,催化XY 结构域进行PIP2的酶促水解,生成三磷酸肌醇(IP3)[7],促进内质网中Ca2+的释放, 进而诱导细胞内 Ca2+振荡的发生[8]。Saunders 等[4]将PLCζ 功能受到抑制的精子注入卵母细胞中,发现Ca2+的振荡不明显,而将完整的PLCζ cRNA 注入小鼠卵母细胞后产生了与正常体外受精相类似的Ca2+振荡现象,表明PLCζ 介导的Ca2+振荡在体外受精过程中发挥重要作用。 研究显示,在牛[9]、鸡[10]和猪[11]等家畜中已经发现PLCζ基因。
体外授精实验结果表明,Plcz1基因缺陷的精子会导致小鼠受精失败,进而导致不育,最近研究发现,在缺失PLCζ 蛋白的情况下,自然交配的小鼠也会导致体内受精产生后代,但产仔数明显下降[12]。因此,自然受精的情况下Plcz1基因缺陷雄鼠是否具有生育能力尚未完全阐明。 本研究利用CRISPR/Cas9 技术构建Plcz1-/-小鼠模型,并对Plcz1-/-小鼠进行扩繁和基因型鉴定,探讨Plcz1基因在雄性小鼠生育过程中的作用,为解决Plcz1基因突变带来的生殖能力障碍提供理论依据。
1 材料和方法
1.1 实验动物
SPF 级C57BL/6 雄鼠6 只,8~12 周龄,体重23~28 g;SPF 级C57BL/6 雌鼠12 只,4~8 周龄,体重14~22 g;SPF 级ICR 雄鼠3 只,10 周龄,体重32~37 g;SPF 级ICR 雌鼠3 只,10 周龄,体重30~35 g,均购自扬州大学比较医学中心[SCXK(苏)2017-0007],实验操作在扬州大学兽医学院兽医大楼的实验室进行[SYXK(苏)2017-0044]。 实验小鼠均饲养于扬州大学比较医学中心的SPF 级动物房,光照周期采用5:00 am~7:00 pm,环境温度控制在20℃~25℃,自由饮水、进食,每周更换1 次垫料。
动物实验伦理审核由扬州大学审批(NSFC2020-SYXY-20),并按照实验动物使用的3R 原则给予人道的关怀。
1.2 主要试剂与仪器
pX330A-1x2、pX330S-2 载体购自Addgene;限制性内切酶BpiⅠ(批号:10020723)、T4 DNA 连接酶(批号:10037474)及限制性内切酶Eco31I(批号:0431712)购自New England Biolabs 公司;2×Es Taq MasterMix(Dye)(批号:01031/50351)、DH5α 感受态细胞(批号:50525)及高纯度质粒小提试剂盒(批号:50433)购自康为世纪生物科技股份有限公司;EndoFree Plasmid Maxi Kit(批号:163013537)购自QIAGEN;普通琼脂糖(批号:EZ6688C178)购自赛国生物科技有限公司;纯化试剂盒(批号:123722)购自 MP Biomedicals; DL2000 marker ( 批 号:AKF0781A)购自TaKaRa Bio;Spe 抗生素(批号:EB15BA0006)购自生工生物工程(上海)股份有限公司。
T100 Thermal Cycler PCR 仪购自美国Bio-Rad公司;ABI PCR 仪miniAmp 及ABI 经典型PCR 仪2720 购自美国Thermo Fisher 公司;天能1600 型凝胶成像+电脑、HE-90 电泳槽购自上海天能生物科技有限公司;恒温水浴锅购自宁波新芝生物科技有限公司;台式冷冻离心机5424R、台式离心机5424、微型离心机MiniSpin 购自德国Eppendorf 公司;ZHTY-50 N 恒温振荡培养箱购自上海知楚仪器有限公司。
1.3 实验方法
1.3.1 设计sgRNA 及sgRNA 双链合成
从NCBI(https:/ /www.ncbi.nlm.nih.gov/)上查询到C57BL/6 小鼠的Plcz1基因的序列(NM_054066.4),由于第一个外显子不存在氨基酸,因此本试验从第二个外显子开始统计外显子数量并标记的。 通过张峰sgRNA 设计的网站(http:/ /crispr.mit.edu/)在第6、7 外显子上选择sgRNA 靶点序列,选择出得分较高的2 条sgRNA,并在其两端添加限制性内切酶BpiⅠ酶切位点,由擎科有限公司合成(如表1 所示)。 其中,如图1 所示的基因敲除示意图,Plcz1-sgRNA6 位于Plcz1基因第6 外显子,Plcz1-sgRNA7 位于Plcz1基因第7 外显子。

表1 Plcz1 基因sgRNA 寡核苷酸链Table 1 SgRNA oligonucleotide chain of Plcz1 gene
将合成的上游引物和下游引物各8 μL、Taq DNA Polymerase Buffer 4 μL 在PCR 管中混合,进行PCR 退火反应:99℃ 10 min,16℃ 10 min。
1.3.2 pX330-sgRNA 重组载体的构建
将退火的寡核苷酸分别插入pX330A 和pX330S 载体,反应体系共2 μL:25 ng/μL pX330A/S 载体0.3 μL,10 μmol/L 退火的寡核苷酸0.5 μL,10×T4 DNA 连接酶缓冲液0.2 μL,BpiI 0.1 μL,T4 DNA 连接酶0.1 μL,无菌蒸馏水0.8 μL。 进行PCR 扩增反应:37℃ 5 min,16℃ 10 min 进行3 个循环,循环反应后,在37℃下进行额外的BpiI 酶消化1 h, 即得到连接产物 pX330A-1x2-sgRNA6 与pX330S-2-sgRNA7,用于后续步骤。
1.3.3 连接产物转化感受态细胞
取冷冻的感受态细胞置于冰浴中,待感受态细胞在冰上融化后,在超净台中向感受态细胞悬液中加入连接产物,用移液器轻轻吹打混匀,置于冰上30 min。 42℃热击90 s,迅速将离心管转移到冰浴中,冰上静置2 min。 在超净台里向每个离心管中加入700 μL 无菌的LB 培养基,不含抗生素,混匀后置于37℃摇床,220 r/min 振荡培养1 h 使菌体复苏。 在超净台中取50 μL 已转化的感受态细胞,加到含Amp 或者Spe 抗生素的LB 固体琼脂培养基上,用无菌的涂布棒将菌液均匀涂开,直至干燥,倒置平板,37℃ 12~16 h 培养。
1.3.4 质粒的提取与鉴定
用灭菌过后的枪头挑取上述培养出的单菌落,接种于5 mL Amp 或Spe 抗生素的LB 液体琼脂培养基置于37℃摇床,220 r/min 振荡培养10~12 h。在超净台中保菌,余下菌液用质粒小提试剂盒提取质粒,取过夜培养的菌液加入离心管(自备)中,13000 r/min 离心30 s 收集菌体沉淀,尽量吸弃上清。 向留有菌体沉淀的离心管中加入250 μL 含RNase A 的Buffer P1,使用涡旋振荡器充分混匀,悬浮菌体沉淀。 加入250 μL Buffer P2,轻轻地上下颠倒混匀6 次,充分混匀使菌体裂解,此时溶液应变得清亮粘稠。 加入350 μL Buffer N3,立即轻柔地上下颠倒混匀10 次,充分混匀,此时应出现白色絮状沉淀。 13000 r/min 离心5 min。 将所得的上清液转移到已装入收集管的吸附柱(Spin Columns DM)中,13000 r/min 离心30 s,倒掉收集管中的废液,将吸附柱重新放回收集管中。 向吸附柱中加入150 μL Buffer PB,13000 r/min 离心30 s。 向吸附柱中加入400 μL Buffer PW,13000 r/min 离心1 min,倒掉收集管中的废液。 将吸附柱置于1 个新的离心管中,向吸附膜的中间部位加入60 μL Buffer EB,室温放置2 min,13000 r/min 离心1 min,将质粒溶液收集到离心管中,-20℃保存质粒。 质粒送至南京擎科生物科技有限公司进行测序,测序引物为通用引物U6-F。
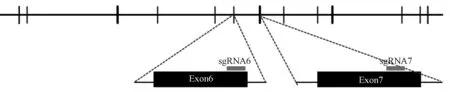
注:Plcz1 基因的外显子由竖线表示。图1 Plcz1 基因的基因结构(NM_054066.4)和CRISPR/CAS9 的靶序列Note.Exons are represented by vertical bars.Figure 1 Gene structure of the mouse Plcz1 gene (NM_054066.4) and target sequences for CRISPR/Cas9
1.3.5 Golden Gate assembly
为构建靶向2 个Plcz1基因组位点的CRISPR/Cas9-核酸酶载体, 将测序正确的pX330A-1x2-sgRNA6 与pX330S-2-sgRNA7 统一到单个载体中。将pX330A/S 载体、Eco31I、T4 DNA 连接酶与10×T4 DNA 连接酶缓冲液在PCR 管中混合,并进行如下热循环反应: 37℃ 5 min,16℃ 10 min 进行25 个循环,循环反应后,在37℃下进行额外的Eco31I 消化1 h,即得到连接产物pX330A-2(如图2 所示),用于后续步骤。
1.3.6 显微注射
测序正确的质粒经纯化后,用注射用水稀释至3 ng/μL,存于-20℃备用。 将注射过PMSG 和hCG的C57BL/6 雌鼠(供体)与C57BL/6 雄鼠合笼,同时将未注射过激素的ICR 雌鼠与结扎的雄鼠合笼,从而获得假孕雌鼠(受体)。 处死超数排卵的供体雌鼠,取出受精卵,使用显微注射仪将质粒注射到受精卵原核中。 麻醉假孕雌鼠,将受精卵移植至受体卵巢内。 3 周后移植成功的受体雌鼠可生出F0代小鼠。
1.3.7 基因敲除小鼠的筛选
提取鼠尾基因组DNA,通过PCR 检测目的片段。 由南京擎科生物科技有限公司合成检测sgRNA 的引物(如表2 所示)。 PCR 反应体系:2×Es Taq MasterMix(Dye)10 μL,F(10 μmol/L)1 μL,R(10 μmol/L)1 μL,DNA(<0.2 μg)1 μL,ddH2O 7 μL。 PCR 反应程序:预变性94℃ 2 min,变性94℃ 30 s、退火60℃ 30 s、延伸72℃ 30 s,循环33 次,终延伸72℃ 2 min。 PCR 扩增产物送至南京擎科生物科技有限公司进行测序, 用SnapGene 软件分析。
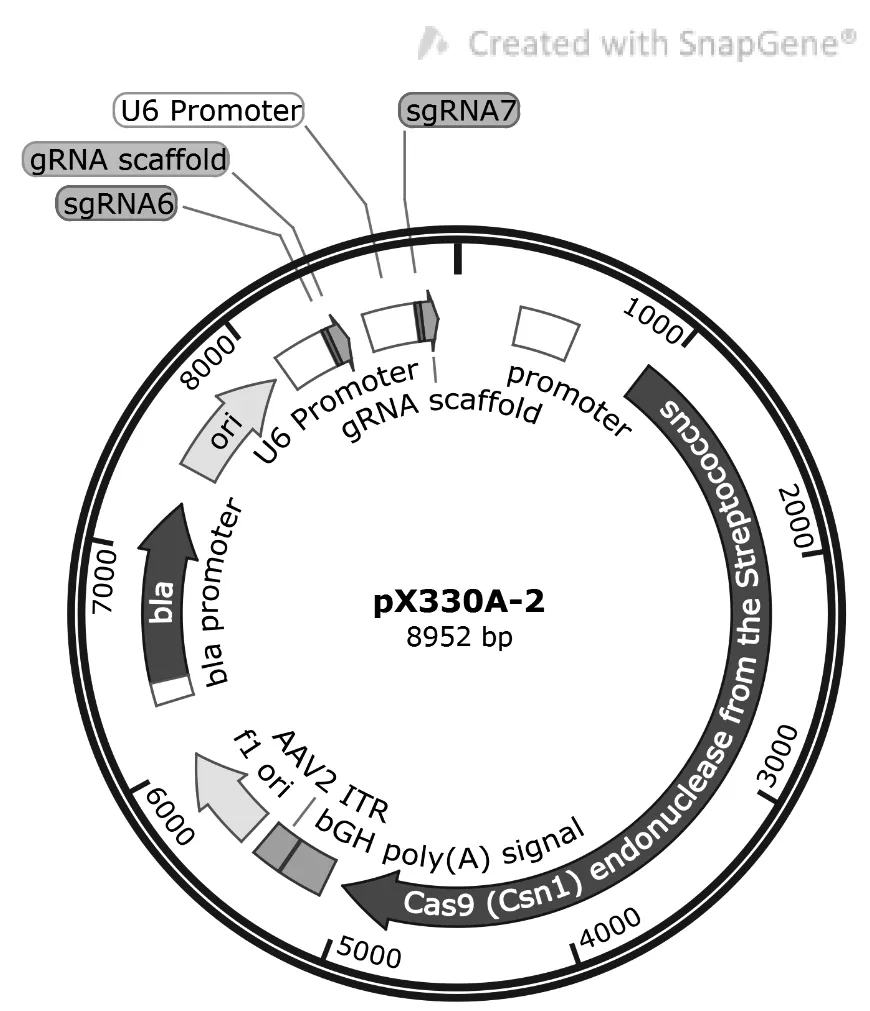
注:图片由SnapGene 提供。图2 pX330A-2 质粒图谱Note.The picture was support by SnapGene software.Figure 2 Plasmid map of pX330A-2

表2 检测目的片段的引物序列Table 2 Primer sequences for detection of target fragment
1.3.8Plcz1-/-雄性小鼠的饲养和繁育
繁育出F0 代小鼠后,与野生型C57BL/6 小鼠合笼,得到F1 代杂合小鼠。 然后将纯合和纯合小鼠、纯合和杂合小鼠、纯合和野生型、杂合和杂合小鼠以及野生型和野生型小鼠等不同基因型间进行合笼,从繁殖情况分析Plcz1基因敲除雄性小鼠的生育能力。
1.4 统计学方法
应用IBM SPSS Statistics 22 统计软件进行统计学分析,组间两两比较采用配对双尾t检验,数据以平均值±标准差(±s)显示。 当P<0.05 时表示差异有统计学意义。
2 结果
2.1 pX330A-sgRNA 重组载体构建
目的序列Plcz1-sgRNA6 和sgRNA7 与载体pX330A 载体连接,通过序列测定验证目的基因,并用SnapGene 软件进行序列比对,结果表明正确插入两对sgRNA 序列(图3)。
2.2 Plcz1 基因敲除小鼠的测序筛选
获得受精卵82 枚,显微注射活胚54 枚,胚胎移植两只ICR 受体(C102、C103),其中C102受体产仔6 只,编号1~6,C103 受体生的8 只后代均被吃。 PCR 扩增包含sgRNA 的片段后进行琼脂糖凝胶电泳(如图4 所示),电泳结果显示:用检测sgRNA6 或sgRNA7 引物进行PCR 扩增,3号小鼠没有电泳条带,其余小鼠电泳条带大小为289 bp;而用检测sgRNA6 上游引物和sgRNA7 下游引物进行PCR 扩增,3 号小鼠在268 bp 处显示电泳条带。 经测序3~4 次鉴定出3、4、5 号小鼠为Plcz1基因敲除小鼠,分别为:(1)Plcz1m3:Plcz1基因第6、7 外显子区域缺失3078 bp,在第295 个氨基酸处开始突变;(2)Plcz1m4:Plcz1基因第7 外显子区域缺失7 bp,在第356 个氨基酸处开始突变,该小鼠在饲养过程中死亡。 (3)Plcz1m5:Plcz1基因第6 外显子区域缺失1 bp,在第295 个氨基酸处开始突变。 同时预测Plcz1m3、Plcz1m4与Plcz1m5的等位基因表达产生的截短蛋白结构域(如图5所示):Plcz1m3和Plcz1m5小鼠的PLCζ 蛋白在X 结构域处产生缺失;Plcz1m4小鼠的PLCζ 蛋白在X-Y连接区产生缺失。
2.3 Plcz1-/-雄性小鼠的生育能力分析
表3 和表4 显示了小鼠Plcz1m3和Plcz1m5后代的生育能力。 结果发现,Plcz1m3和Plcz1m5雄鼠与野生型雌鼠交配虽然可以产生后代,但与野生型雄鼠(8.5±1.4)相比,后代数量极显著性降低(Plcz1m3:2.5±0.5;Plcz1m5:2.3±1.1)。 同时与野生型雄鼠相比(受精率100%;后代死亡率4.7%),Plcz1m3雄鼠受精率13.3%;Plcz1m5雄鼠受精率50%,后代死亡率24%。Plcz1m3雄鼠和Plcz1m3雌鼠交配,后代数量极显著性降低(3.0±2.2),Plcz1m3雄鼠受精率30%,后代死亡率高达100%,而Plcz1m5雄鼠与Plcz1m5雌鼠交配不产生后代。 由此可见,Plcz1m3和Plcz1m5雄鼠可以和不同基因型雌鼠进行交配,但其受精率、出生率以及死亡率均显著性下降。 结果表明,Plcz1-/-雄鼠自然交配可以产生后代,但是生育能力显著性下降,同时Plcz1m5雄鼠较Plcz1m3雄鼠受精率高,但后代死亡率也高。
表4 小鼠Plcz1m5 后代生育能力参数(±s)Table 4 Fertility parameters of mouse Plcz1m5 offspring
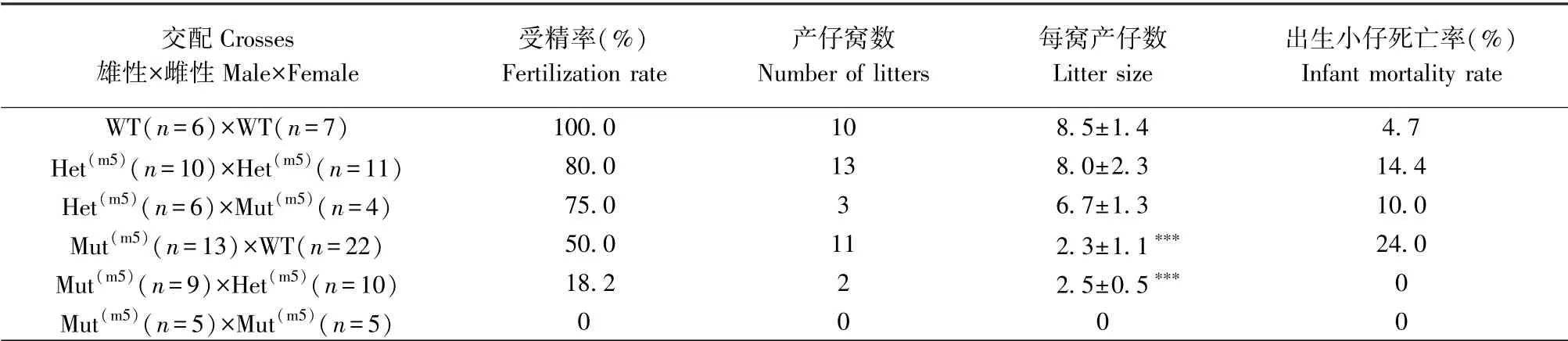
表4 小鼠Plcz1m5 后代生育能力参数(±s)Table 4 Fertility parameters of mouse Plcz1m5 offspring
注:杂合:Plcz1+/m3 或Plcz1+/m5;纯合:纯合子Plcz1m3 或 Plcz1m5;WT:野生型;雄性和雌性小鼠之间根据指定的基因型建立交配对。 与野生型小鼠每窝产仔数相比,∗∗∗P<0.001。Note.Het, Plcz1+/m3 or Plcz1+/m5.Mut, homozygote Plcz1m3 or Plcz1m5.WT, Wild-type.Mating pairs were set up between males and females with genotypes as indicated.Compared with the litter size of wild-type mouse,∗∗∗P<0.001.
交配Crosses雄性×雌性Male×Female受精率(%)Fertilization rate产仔窝数Number of litters 每窝产仔数Litter size出生小仔死亡率(%)Infant mortality rate WT(n=6)×WT(n=7) 100.0 10 8.5±1.4 4.7 Het(m5)(n=10)×Het(m5)(n=11) 80.0 13 8.0±2.3 14.4 Het(m5)(n=6)×Mut(m5)(n=4) 75.0 3 6.7±1.3 10.0 Mut(m5)(n=13)×WT(n=22) 50.0 11 2.3±1.1∗∗∗ 24.0 Mut(m5)(n=9)×Het(m5)(n=10) 18.2 2 2.5±0.5∗∗∗ 0 Mut(m5)(n=5)×Mut(m5)(n=5) 0 0 0 0
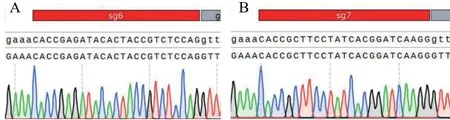
注:A:sgRNA6 的测序结果与预测构建的载体的序列比对;B:sgRNA7 的测序结果与预测构建的载体的序列比对。图3 载体测序对比图Note.A, Sequencing results of sgRNA6 were compared with the predicted vector.B, Sequencing results of sgRNA7 were compared with the predicted vector.Figure 3 Comparison of vector sequencing
表3 小鼠Plcz1m3 后代生育能力参数(±s)Table 3 Fertility parameters of mouse Plcz1m3 offspring
表3 小鼠Plcz1m3 后代生育能力参数(±s)Table 3 Fertility parameters of mouse Plcz1m3 offspring
注:与野生型小鼠每窝产仔数相比, ∗P<0.05,∗∗∗P<0.001。Note.Compared with the litter size of wild-type mouse, ∗P<0.05,∗∗∗P<0.001.
交配Crosses雄性×雌性Male × Female受精率(%)Fertilization rate产仔窝数Number of litters每窝产仔数Litter size出生小仔死亡率(%)Infant mortality rate WT(n=6)×WT(n=7) 100.0 10 8.5±1.4 4.7 Het(m3)(n=16)×Het(m3)(n=15) 75.0 25 6.3±1.5∗∗∗ 21.0 Het(m3)(n=6)×Mut(m3)(n=7) 55.6 9 6.9±1.5∗ 16.1 Mut(m3)(n=15)×WT(n=15) 13.3 2 2.5±0.5∗∗∗ 0 Mut(m3)(n=17)×Het(m3)(n=17) 17.7 3 3.0±0.8∗∗∗ 0 Mut(m3)(n=10)×Mut(m3)(n=6) 30.0 3 3.0±2.2∗∗∗ 100.0
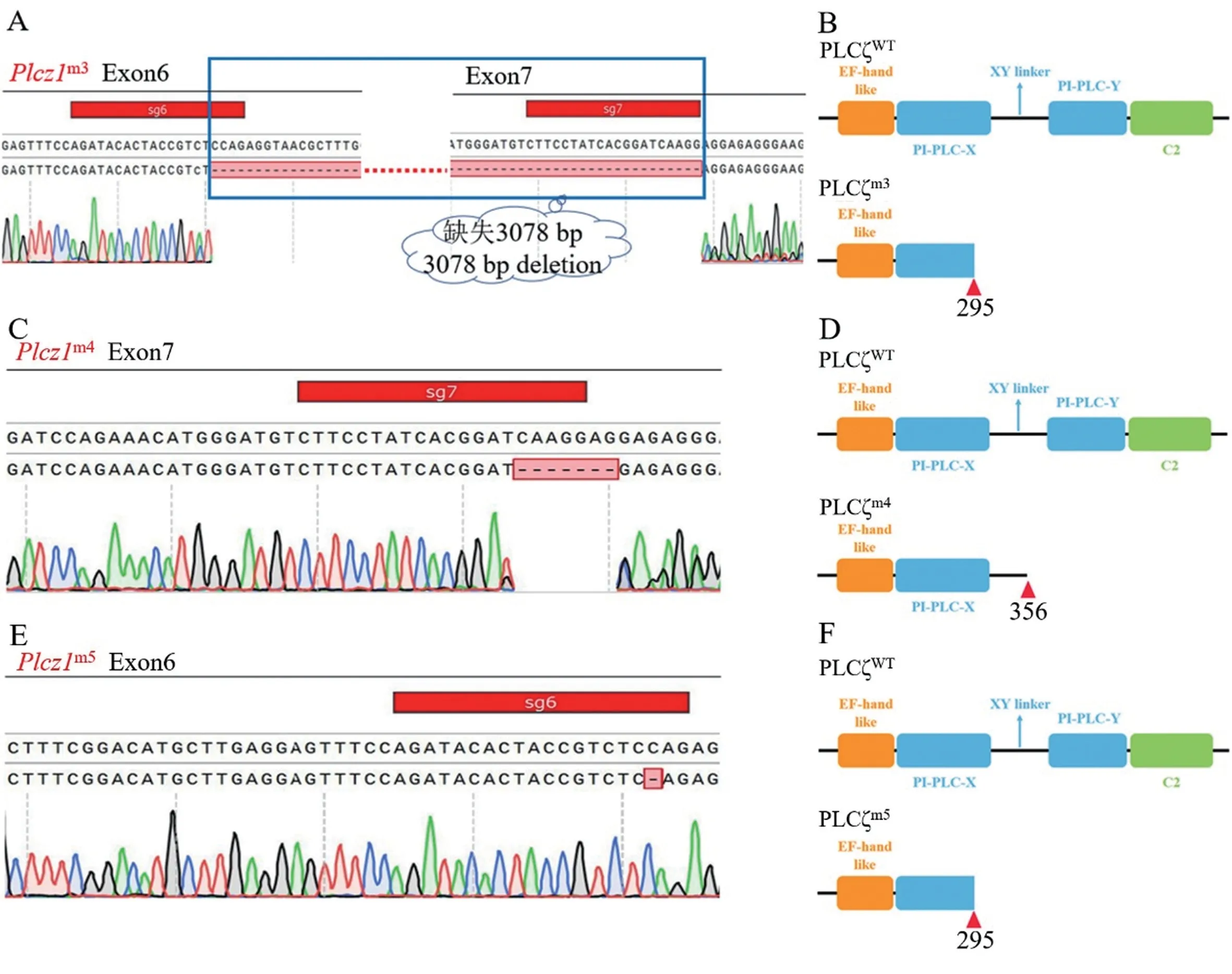
注:A、C、E:野生型Plcz1(Plcz1WT)等位基因和3 个核苷酸缺失突变Plcz1 等位基因的基因组序列比较,虚线标红为缺失序列;B、D、F:预测了野生型小鼠PLCζ 蛋白(PLCζWT)和突变的PLCζm3(B)、PLCζm4(D)以及PLCζm5(F)等位基因表达的截短蛋白的结构域。图5 小鼠Plcz1m3、Plcz1m4 以及Plcz1m5 突变等位基因的表达分析Note.A, C, E, Comparison of genomic sequences from wild-type Plcz1(Plcz1WT) allele and three mutant Plcz1 alleles harbouring nucleotide deletions, the missing sequence is marked red by a dotted line.B, D, F, Predicted protein-domain structures for wild-type mouse PLCζ protein (PLCζWT) and truncated proteins resulting from expression of mutant PLCζm3(B), PLCζm4(D) and PLCζm5(F) alleles.Figure 5 Expression analysis of mouse Plcz1m3, Plcz1m4and Plcz1m5mutant alleles
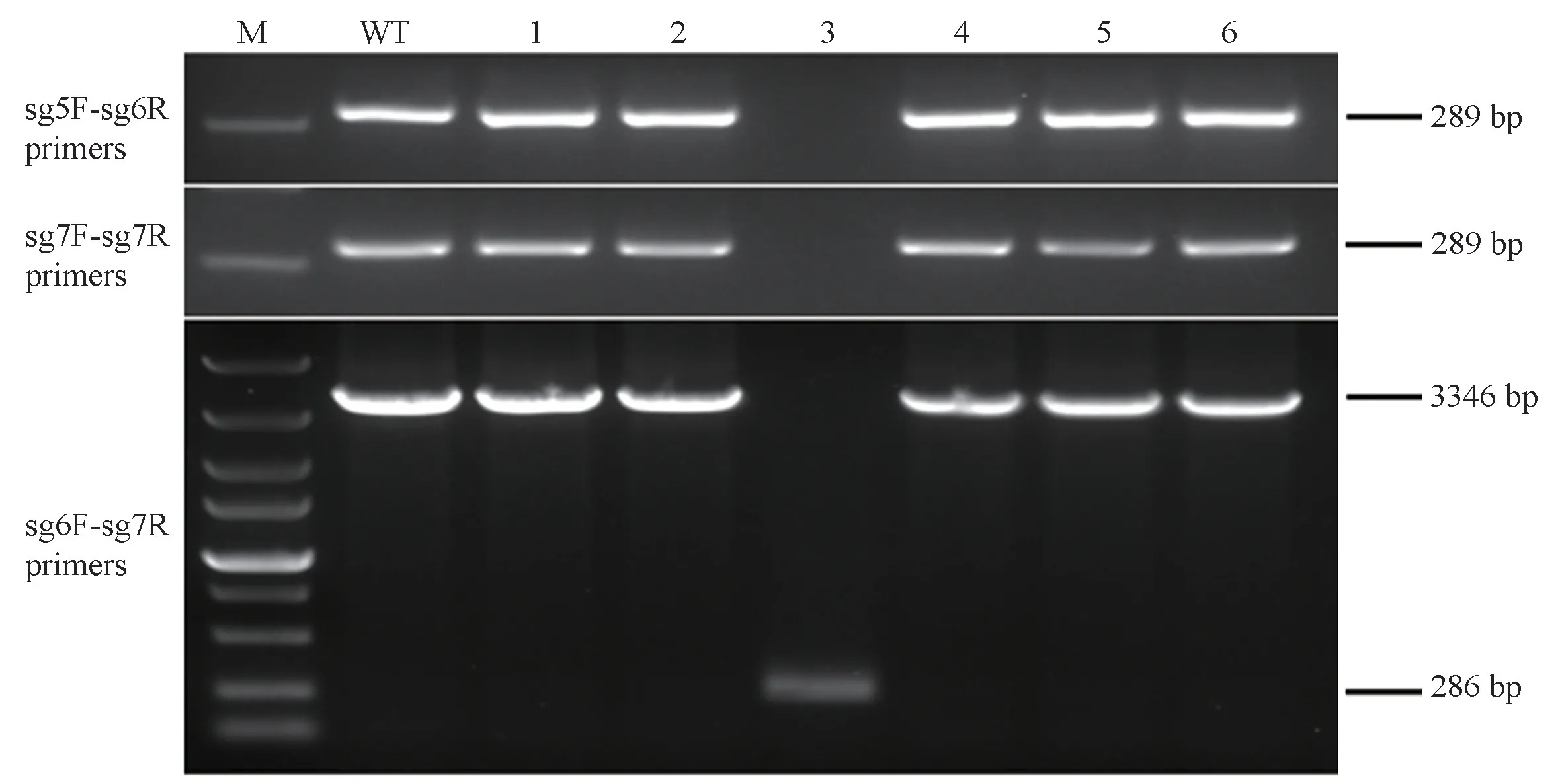
注:M:DL5000 Marker;WT:野生型;1~6:原代小鼠的鼠尾DNA。图4 PCR 法鉴定小鼠Plcz1 基因突变位点Note.M, DL5000 Marker.WT, Wild type.1~6, Tail DNA of primary mice.Figure 4 Identification of mouse Plcz1 gene mutation site by PCR
3 讨论
不孕症是一种影响男性和女性的复杂和多因素的疾病,全球临床数据显示,每7 对夫妇中就有1 对经历不孕不育[13]。 即使采用胞浆内单精子注射(ICSI),高达5%的ICSI 治疗周期仍然失败,仅在英国每年就有至少1000 对夫妇受到影响[14]。 目前,卵子激活过程中的缺陷被认为是这一失败的主要来源[15]。 许多临床报告已将人类PLCζ 缺陷与卵母细胞激活失败联系在一起,同时某些类型的男性不育与人类精子中PLCζ 的异常表达、功能、结构以及定位密切相关[16]。 因此,本研究构建的Plcz1基因敲除小鼠模型对研究PLCζ 在精子和卵母细胞中的表达和功能活性的调控机制,以及找出不孕症的潜在治疗方案具有临床意义。
利用基因组靶向编辑技术制备动物模型是当今研究的热点领域之一。 CRISPR/Cas9 系统使用RNA 引导的核酸酶切割外源基因[17],相较于其他基因编辑技术,具有易于定制、更高的靶向效率以及促进多重基因组编辑的能力[18-19],操作更加简单,成本较低[20]。 Sakuma 等[21]用CRISPR/Cas9 构建的系统为多重基因组/表观基因组编辑、同时激活/抑制多个基因等提供了有效的靶向策略。Golden Gate 载体构建方法,使构建质粒更加便利[22]。 为保证CRISPR/Cas9 的切割效率,本研究选择脱靶位点较少、sgRNA 与其互补序列错配率也较少的靶点,且得分较高的靶点,同时也控制CRISPR/Cas9 的剂量,因为剂量也是影响脱靶突变的另一个因素,最大限度地减少脱靶突变。 此外,通过利用Cas9 与多个sgRNA 一起使用的优势,可成功地实现sgRNA 靶点之间的大片段缺失或倒置[23]。
利用显微注射方法将质粒注射到胚胎原核中[24],21 d 后,6 只F0 代小鼠出生,鼠尾DNA 经过PCR 后琼脂糖凝胶电泳和测序分析,证明3 只小鼠有Plcz1基因缺失,成功构建出Plcz1基因敲除小鼠模型。 野生型或Plcz1-/-雄性小鼠分别与野生型雌性小鼠交配,结果表明Plcz1-/-雄性小鼠可以繁育后代,但是与野生型雄性小鼠相比其产仔数量显著性下降,此结果与Hachem 等[12]的研究结果相符,为阐明Plcz1基因突变带来的生殖能力障碍提供理论依据。
目前,研究者越来越重视卵母细胞激活途径的选择与优化,在体外激活卵母细胞的过程中,研究者往往通过选取不同的激活方式获得动物的胚胎,而卵母细胞的人工激活效率会直接影响胚胎的发育质量。 受精时,PLCζ 作为精子中一种重要的因子在激活卵母细胞第二次减数分裂过程中发挥重要作用。 精卵融合后,PLCζ 通过精子进入卵质,并催化PIP2水解生成IP3。 IP3触发的细胞内Ca2+释放,产生特有的胞质Ca2+振荡,导致卵子激活,从而启动胚胎发育过程。 Nomikos 等[25]证明在表达功能障碍的PLCζ 不能激活的卵母细胞中显微注射重组人PLCζ,可以有效地挽救失败的激活,并正常发育至囊胚阶段。 在人类生殖过程中,PLCζ 蛋白缺陷的男性会不育[26-27]。 Hachem 等[12]首次在哺乳动物上证明,在没有卵母细胞激活的情况下,自然交配和受精可以产生后代,与本文研究结果相符,相较于野生型雄鼠,Plcz1-/-雄鼠在受精率和繁殖率上均有显著性下降,是否存在别的途径激活小鼠体内的卵母细胞还有待商榷,需要对Plcz1基因敲除小鼠进行深入的机制研究。 同时,Dai 等[28]证明催化结构域变异的精子会受到损害,失去水解PIP2的功能,导致异常的Ca2+振荡,不能激活卵母细胞使受精失败。 Unnikrishnan 等[29]表明PLCζ 在精子中的分布在不同物种中存在差异:在人类精子头部的顶体、赤道段和顶体后区域,以及尾部区域;在小鼠精子的顶体和顶体后区域。 但是,PLCζ 的不同结构区域调节PLCζ 定位的机制,以及对于精子获能的具体作用尚不清楚。
本研究利用CRISPR/Cas9 系统构建的Plcz1基因敲除(Plcz1-/-)小鼠模型,为进一步揭示Plcz1基因缺失导致的生殖障碍发病机制提供理论依据,同时为治疗生育能力障碍等问题的研究提供了有效的动物模型。 但是关于PLCζ 的确切作用机制及其在卵母细胞激活过程中的作用程度仍然存在许多问题,所以需要进一步探索。
