CO2 加氢制甲酸理论研究及高效铁基催化剂设计
2022-08-13胡兴邦
刘 聪, 胡兴邦
(1. 山东国邦药业股份有限公司, 山东 潍坊 261108; 2. 南京大学 化学化工学院, 江苏 南京 210023)
二氧化碳(CO2)是一种主要温室气体. 近年来,大气中的CO2浓度正快速增加. 为此, 我国明确提出了“碳达峰”和“碳中和”目标. 使用CO2为原料合成有用的化工产品是碳减排的有效途径. 越来越多的学术和产业界研究人员开始关注CO2的利用问题[1].
在文献报道的CO2利用方法中, CO2加氢制甲酸是最吸引人的过程之一[2-4]. 一方面, 甲酸在工业上被广泛使用. 另一方面, CO2加氢制甲酸具有100%的原子经济性. CO2加氢包括两个步骤: 氢气(H2)活化形成金属-氢(M-H)键以及CO2插入M-H 键[3-16]. 这一反应由于需同时活化惰性的CO2和H2而充满挑战. 开发高活性催化剂对于实现CO2加氢制甲酸极为重要. 文献中报道的CO2加氢制甲酸催化剂包括贵金属(如Ir[17-18]、 Rh[19-20]、Ru[21-22]、 Pt[23]、 Pd[24-25]和Re[26])和非贵金属(如Cu[27-28]、 Mn[29-30]、 Fe[31-34]、 Co[35-36]和 Mo[37])催化剂. 在这些催化剂中, 贵金属催化剂展现出了很高的催化活性, 而非贵金属催化剂活性较低. 比如,基于Ir 的tBuPNP-Ir(III)催化剂的每摩尔催化剂单位活性中心上底物的转化数(TON)高达3 500 000[17].同样, 基于Ru 的tBuPNP-Ru 催化剂的TON 达到6 000 000[21]. 相对而言, 非贵金属催化剂的活性要低很多. 基于Co 的Co(dmpe)2H 代表了活性最高的CO2加氢制甲酸非贵金属催化剂, 但其TON 只有9400[35], 比tBuPNP-Ir(III)和tBuPNP-Ru 的TON分别低了372[17]和638[21]倍.
尽管贵金属催化剂的催化活性非常优异, 但昂贵的价格严重限制了其大规模工业化应用. 铁(Fe)是地球上含量最丰富的过度金属元素之一. 然而,文献报道的CO2加氢制甲酸铁基催化剂活性都非常低. 比如, 基于Fe 的PNP-Fe(III)的TON 只有788[31].
PNP 型金属配合物在CO2加氢制甲酸过程中被广泛使用[17,19,21,31]. 作为课题组在CO2加氢领域工作的重要组成部分[28,38-39], 我们对基于Fe 的PNPFe 化合物进行不同官能化修饰理论研究, 期望从理论上揭示能大幅提升PNP-Fe 催化CO2加氢制甲酸活性的方法, 为高效CO2加氢制甲酸催化剂开发提供理论基础.
1 计算方法
众所周知, 密度泛函理论(DFT)在给出合理计算结果的同时具有较高的计算效率. 基于DFT的B3LYP 方法已经被广泛用于贵金属和非贵金属催化的CO2加氢制甲酸反应[6-7,10,16,38-39], 并给出了可与实验相佐证的计算结果[6,16,38-39]. 因此, 我们采用Gaussian09 程序所包含的B3LYP 方法进行计算. 对于体系中的金属原子, 使用LANL2DZ 基组.对于除金属外的其它原子, 使用6-311+G*基组(后文简写为B3LYP/LANL2DZ/6-311+G*). 计算中使用EmpiricalDispersion= GD3BJ 关键词进行色散校正. 所有的结构优化、 能量计算以及零点能校正都采用以上所述计算方法. 计算所得过渡态均有且只有一个虚频. 由于CO2加氢制甲酸常常在四氢呋喃中进行[28,38-39], 因此我们进一步采用PCM 溶剂模型、 使用UFF 原子半径、 以四氢呋喃为溶剂, 对所有优化构型进行了能量计算以及零点能校正(溶剂效应修正采用气态优化所得构型). 热力学修正的温度和压力分别是25.15 ℃和0.10 MPa.
计算同时考虑了过渡金属的高、 低两种不同自旋态. 对于PNP-M(M=Fe, Ru)而言, 低自旋态化合物(自旋多重度=1)远比高自旋态(自旋多重度=3)的要稳定. 比如, 高自旋态PNP-Ru 的吉布斯自由能比低自旋态的要高195.3 kJ/mol. 高自旋态PNP1-Fe 的吉布斯自由能比低自旋态的要高165.5 kJ/mol. 因此, 后续主要关注更加稳定的低自旋态PNP-M 催化剂及其催化的反应过程. 同时, 由于催化剂自旋态能量差远高于反应最大活化能能垒高度, 因此反应过程不存在自旋交叉. 文献中采用Fe或Ru 类催化剂催化CO2加氢制甲酸的理论计算,同样也没有自旋交叉存在[9,13,16].
2 结果与讨论
在迄今文献报道的CO2加氢制甲酸催化剂中,tBuPNP-Ru 具有最高的催化活性(TON=6 000 000[21]).因此, 我们首先对tBuPNP-Ru 催化的CO2加氢制甲酸过程进行研究, 相应活化能数据可以作为标准来评估进一步设计的PNP-Fe 催化剂活性. 结合文献对CO2加氢制甲酸的理论计算结果[3-16], 我们采用图1 所示催化循环机制: 首先CO2插入M-H 键形成M-HCO2中间体(Int1), 然后Int1 上的HCO2阴离子旋转形成更加稳定的M-OCOH中间体(Int2),接下来活化氢气形成催化剂-甲酸配合物(Pro), 最后在碱的作用下催化剂得以还原并生成甲酸盐.
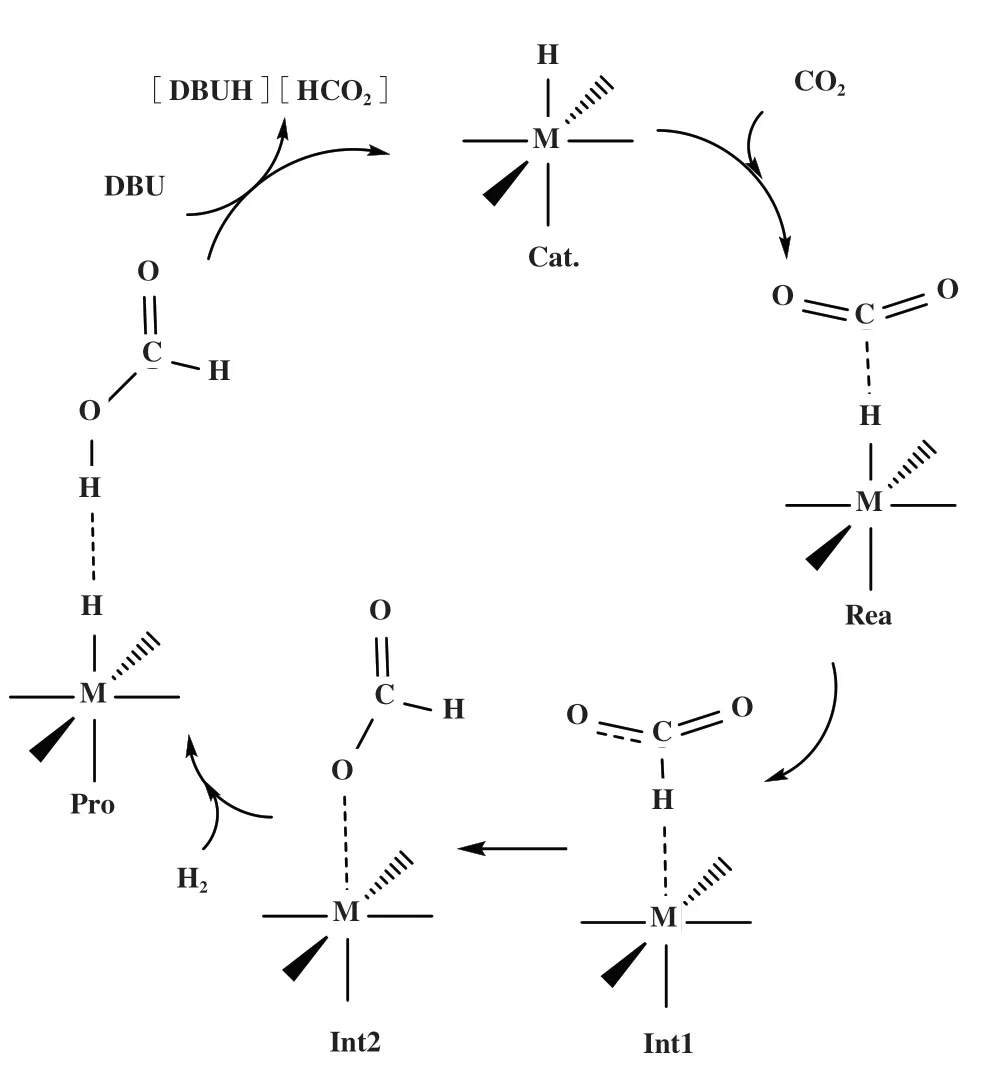
图1 CO2加氢制甲酸催化循环(Rea→Int2: CO2插入; Int2→Pro: H2活化)Fig.1 The catalytic process of CO2 hydrogenation to formic acid(Rea→Int2: CO2 insertion; Int2→Pro: H2 activation)
图2 展示了PNP-Ru 催化CO2加氢制甲酸过程的CO2插入Ru-H 键以及H2活化形成Ru-H键等步骤的吉布斯自由能变化. 图3 展示了PNPRu催化的CO2加氢制甲酸反应物、 过渡态、 中间体、 产物的优化构型. Ru-H键上的氢可经由过渡态TS1PNP-Ru从活性中心Ru向CO2迁移, 相应活化能只有30.5 kJ/mol. 之后, 形成包含HCO2基团的中间体Int1PNP-Ru. HCO2基团可经由TS2PNP-Ru过渡态发生旋转形成中间体PNP-Ru-OCOH (Int2PNP-Ru).紧接着, 通过过渡态TS3PNP-Ru对H2进行活化. H2与Int2PNP-Ru结合为吸热过程, 过渡态TS3PNP-Ru中HH 键 长 为0.093 nm, 与Rh(PH3)2(0.110 nm)[3]和石墨烯负载Cu (0.101 nm)[11]催化的过程十分相似. 活化H2的过渡态TS3PNP-Ru的能垒为76.1 kJ/mol,这一较低的能垒使得PNP-Ru成为目前实验上观察到活性最高的CO2加氢制甲酸催化剂之一[21].
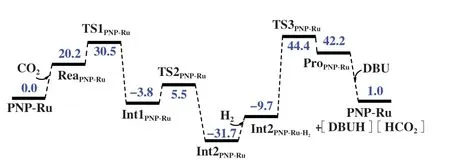
图2 含CO2、 H2结合过程的PNP-Ru催化的CO2加氢制甲酸能量图Fig.2 The energy diagram of CO2 hydrogenation to formic acid catalyzed by PNP-Ru including the binding processes of CO2 and H2(the shown values are Gibbs free energy in kJ/mol, calculation method: B3LYP/LANL2DZ/6-311+G*; energy barrier for CO2 insertion and H2 activation are 30.5 and 76.1 kJ/mol respectively)
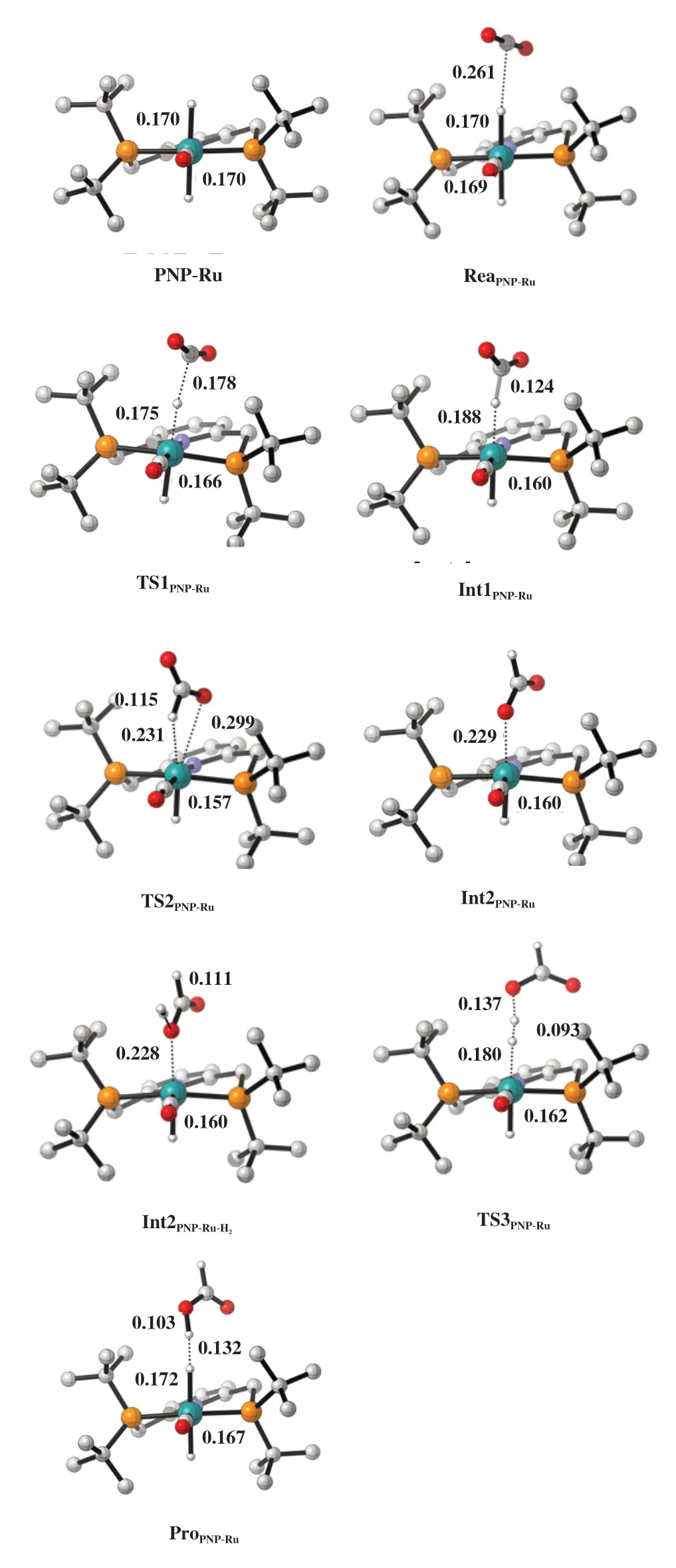
图3 含CO2、 H2结合过程的PNP-Ru催化的CO2加氢制甲酸反应物、 过渡态、 中间体、 产物的优化构型Fig.3 The optimized structures of the reactant, transition states,and intermediates in the CO2 hydrogenation to formic acid catalyzed by PNP-Ru including the binding processes of CO2 and H2(The shown values are bond length in nm, calculation method:B3LYP/LANL2DZ/6-311+G*)
经由TS3PNP-Ru可以得到PNP-Ru-甲酸配合物.研究已经表明CO2加氢制甲酸过程在热力学上不利[40], 因此往往需额外添加有机或无机碱来推动反应进行[17-37], 即通过碱使甲酸从催化剂上脱落, 形成甲酸盐, 同时催化剂得以恢复. 1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)是CO2加氢制甲酸常采用的碱, 在DBU 存在下, 催化循环总的吉布斯自由能变是1.0 kJ/mol(图2). 由于吉布斯自由能为微小的正值, 所以文献中常常采用加入过量碱的方法来推动反应进一步进行[17-37].
为了研究配体修饰对铁基催化剂催化活性的影响并发现活性更高的CO2加氢制甲酸催化剂, 我们一共研究了12 种具有不同结构的Fe 基催化剂(图4). 这些催化剂包括在PNP 配体上进行不同吸电、给 电 修 饰 的 结 构(PNP1~PNP6、 PNP9 和PNP10),包含及不包含分子内氢键的结构(PNP7、 PNP8 和PPP2). 此外, 还将PNP配体吡啶环上的N原子替换成P原子(PPP1和PPP2).
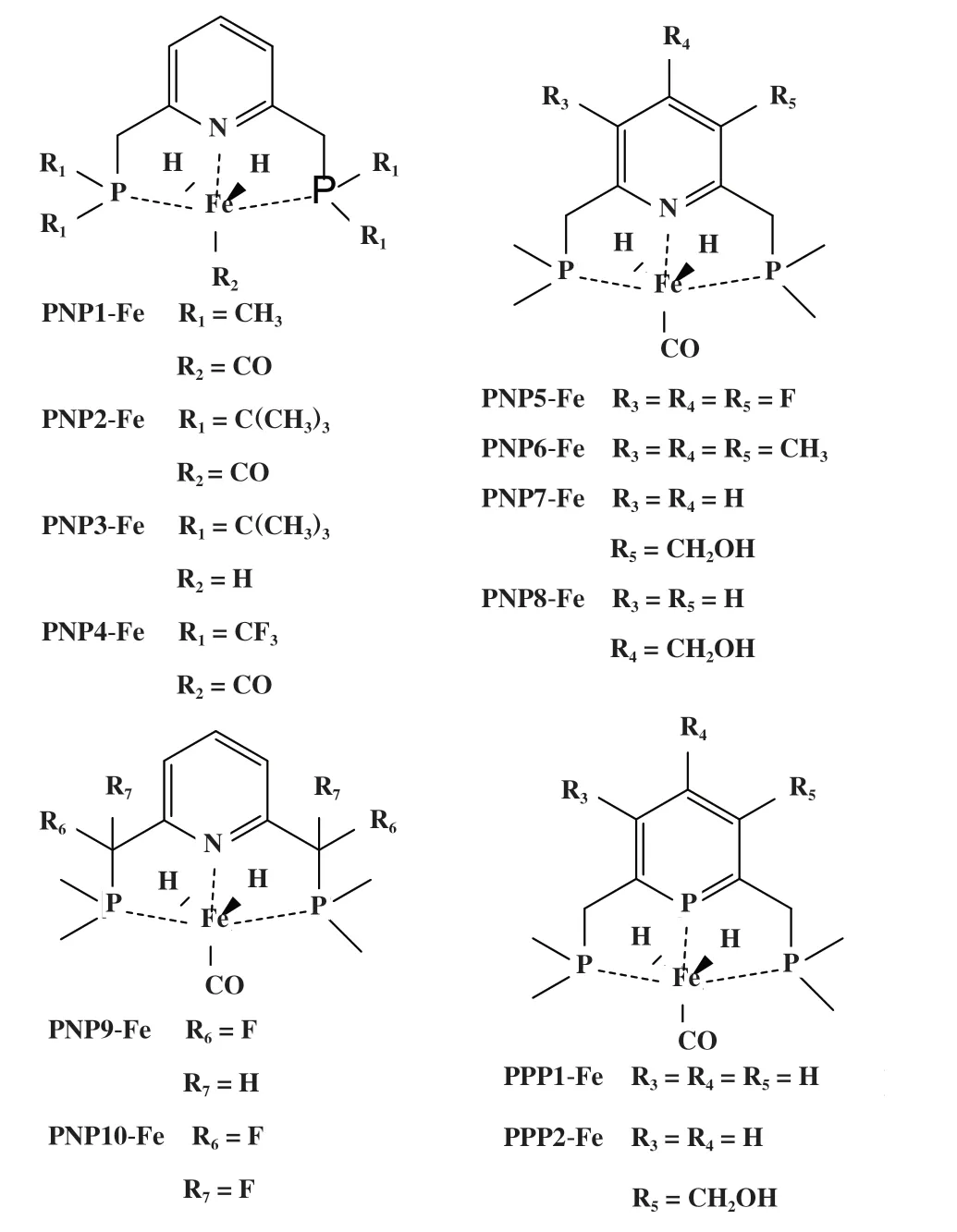
图4 铁基催化剂结构Fig.4 The structures of Fe-based catalysts
图5 和图6 展示了PNP1-Fe 催化CO2加氢制甲酸过程的H2活化形成Fe-H 键以及CO2插入Fe-H 键等步骤的能量图及优化构型. Fe-H 键上的氢可经由过渡态TS1PNP1-Fe向CO2迁移, 相应过渡态能垒只有55.2 kJ/mol. H2活化也是PNP1-Fe 催化过程的速控步骤, 相应过渡态能垒为99.9 kJ/mol.这一H2活化能垒比使用PNP-Ru 为催化剂时的高出23.8 kJ/mol, 这使得PNP1-Fe 具有相对较低活性.
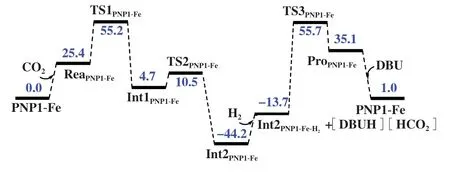
图5 含CO2、 H2结合过程的PNP1-Fe催化的CO2加氢制甲酸能量图Fig.5 The energy diagram of CO2 hydrogenation to formic acid catalyzed by PNP1-Fe including the binding processes of CO2 and H2(The shown values are Gibbs free energy in kJ/mol, calculation method: B3LYP/LANL2DZ/6-311+G*; energy barrier for CO2 insertion and H2 activation are 55.2 and 99.9 kJ/mol respectively)

图6 含CO2、 H2结合过程的PNP1-Fe催化的CO2加氢制甲酸过渡态的优化构型Fig.6 The optimized structures of transition states in the CO2 hydrogenation to formic acid catalyzed by PNP1-Fe including the binding processes of CO2 and H2(The shown values are bond length in nm, calculation method: B3LYP/LANL2DZ/6-311+G*)
由于H2活化是CO2加氢制甲酸过程的速控步骤, 我们还研究了其他修饰方式的PNP-Fe 催化剂催化H2的活化过程. 图7 展示了不同修饰PNP-Fe催化CO2加氢制甲酸过程的H2活化形成Fe-H 键的过渡态优化构型, 对应的能垒展示于表1中. 将PNP-Fe 上 和Fe 配 位 的CO 替 换 为H 原 子 会 使H2活化能垒增加19.1 kJ/mol (TS3PNP2-Fevs. TS3PNP3-Fe).在PNP配体上进行F原子修饰也会显著增加H2活化能垒. 比如, 将P原子上的-CH3修饰为-CF3使H2活化能垒增加37.9 kJ/mol. 将吡啶环上的H原子进行F原子取代后, H2活化能垒增加了21.3 kJ/mol(TS3PNP5-Fevs. TS3PNP1-Fe). 在亚甲基上进行F原子取代也给出类似的结果(TS3PNP9-Fe和TS3PNP10-Fe). 在P原子上进行-CH3取代, 有利于中间体Int2 和H2的结合, 从而使得反应活化能下降, 比如TS3PNP2-Fe的活化能比TS3PNP1-Fe的低47.7 kJ/mol. 引入分子内氢键可稳定H2活化过渡态TS3, 从而降低过渡态能垒. 比如, TS3PNP7-Fe的能垒比TS3PNP1-Fe的要低8.7 kJ/mol. 而当分子内氢键比较弱时, 其对降低活化能的贡献也相应变弱(TS3PNP7-Fe和TS3PNP8-Fe). 值得注意的是, 将PNP配体吡啶环上的N原子替换为P原子能显著降低H2活化能垒14.3 kJ/mol (TS3PPP1-Fevs. TS3PNP1-Fe).
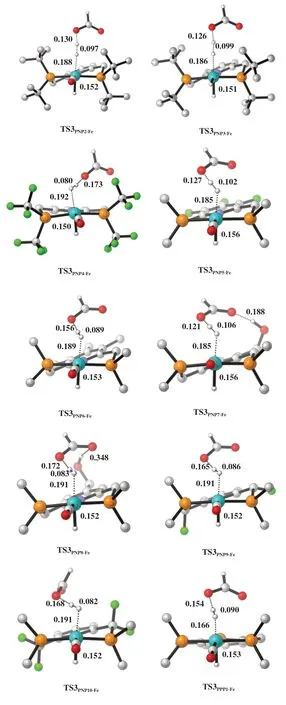
图7 使用不同PNP-Fe催化剂速控步骤过渡态的优化构型Fig.7 The optimized transition states using different Fe-based catalysts (The values are bond length in nm, calculation method:B3LYP/LANL2DZ/6-311+G*)
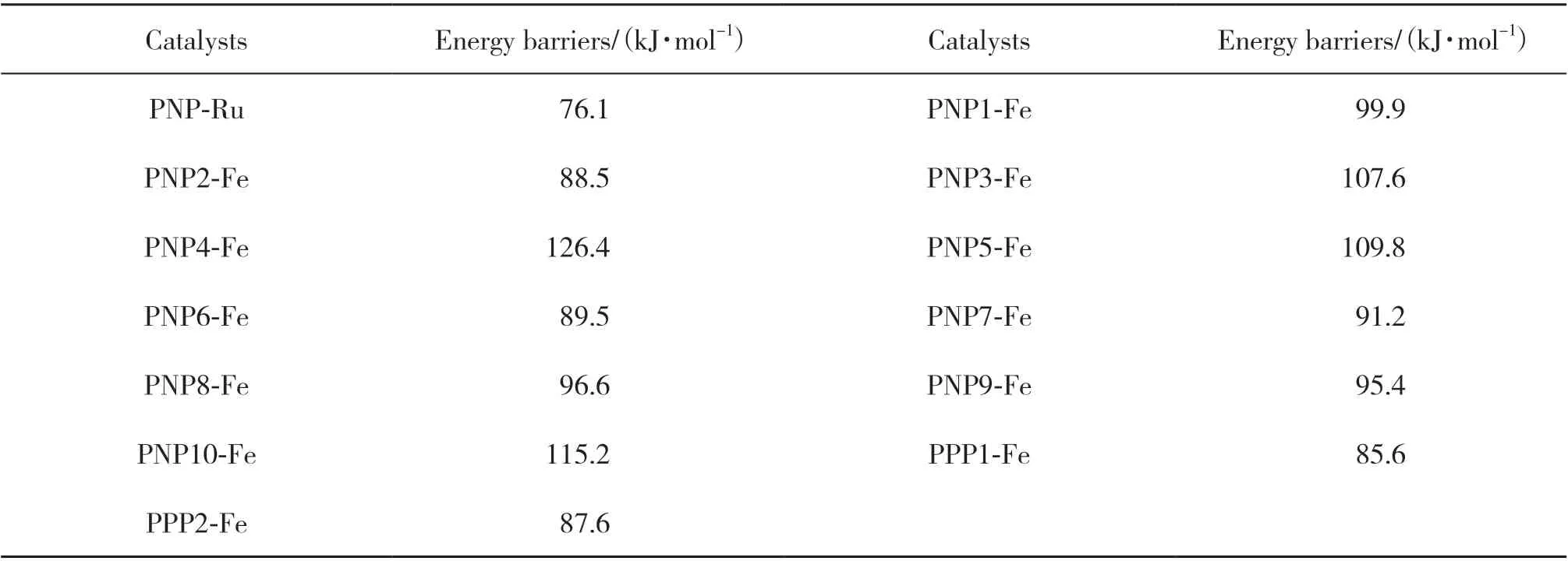
表1 不同催化剂速控步骤活化能垒Table 1 The energy barriers of rate-determining step in the presence of different catalysts
基于以上发现, 我们设计了一个包含分子内氢键及P 原子取代的催化剂(PPP2-Fe). 希望通过P原子取代来调控活性中心金属Fe 的电子结构、 同时通过分子内氢键来稳定过渡态, 从而获得更高活性的CO2加氢制甲酸催化剂. 图8 展示了PPP2-Fe催化CO2加氢制甲酸过程的H2活化形成Fe-H 键以及CO2插入Fe-H键两个步骤的能量图. 图9展示了PPP2-Fe催化的CO2加氢制甲酸反应物、 过渡态、 中间体、 产物的优化构型. 非常值得注意的是,使用PPP2-Fe进行H2活化, 相应活化能垒为87.6 kJ/mol, 比不含氢键的PPP1-Fe体系的能垒稍高2.0 kJ/mol. 虽然氢键形成可在一定程度上稳定过渡态, 通过对比TS3PPP2-Fe和TS3PPP1-Fe的结构可以发现, 对于P取代的催化体系而言, 氢键形成过程会引起芳环结构一定扭曲, 从而抵消了氢键的稳定贡献. 当使用PPP2-Fe为催化剂时, CO2插入Fe-H键也很容易发生, 相应过程能垒只有28.4 kJ/mol (TS1PPP2-Fe).
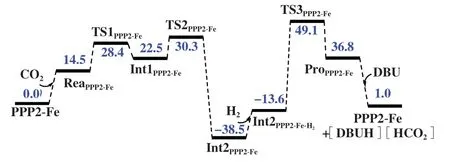
图8 含CO2、 H2结合过程的PPP2-Fe催化的CO2加氢制甲酸能量图Fig.8 The energy diagram of CO2 hydrogenation to formic acid catalyzed by PPP2-Fe including the binding processes of CO2 and H2 (The shown values are Gibbs free energy in kJ/mol, calculation method: B3LYP/LANL2DZ/6-311+G*; energy barrier for CO2 insertion and H2 activation are 30.3 and 87.6 kJ/mol respectively)
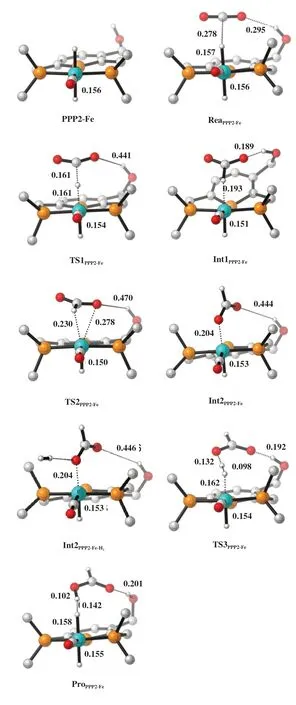
图9 含CO2、 H2结合过程的PPP2-Fe催化的CO2加氢制甲酸反应物、 过渡态、 中间体、 产物的优化构型Fig.9 The optimized structures of the reactant, transition states,and intermediates in the CO2 hydrogenation to formic acid catalyzed by PPP2-Fe including the binding processes of CO2 and H2(The shown values are bond length in nm, calculation method: B3LYP/LANL2DZ/6-311+G*)
PPP1-Fe 是我们研究的不同铁基催化剂中具有最低速控步骤能垒的一个, 其速控步骤能垒为357.8 kJ/mol, 催化活性与贵金属比较接近. 同时, 这一能垒比我们研究的速控步骤能垒最高的PNP4-Fe降低了170.5 kJ/mol, 显示了配体良好的调控催化活性能力(表1). 以上结果为实验开发用于CO2加氢制甲酸的高活性铁基催化剂提供了良好的借鉴和理论基础.
3 结论
为了探索用于CO2加氢制甲酸的高活性铁基催化剂, 采用理论计算的方法, 系统研究了PNPRu 以及12 种不同PNP-Fe 催化剂催化的CO2加氢制甲酸过程. 研究发现: CO2加氢制甲酸包括H2活化及CO2插入金属氢键两个步骤, H2活化是整个反应的速控步骤. 铁基催化剂活性普遍低于Ru 催化剂, 但对PNP 配体进行合理修饰可显著降低两种活性差异. PNP 配体上的F 原子取代会降低催化剂活性, 而提供分子内氢键以及吡啶环上的P 原子取代可增加催化剂活性. 我们设计的铁基催化剂中,PPP1-Fe 具有最高活性, 其催化CO2加氢制甲酸过程速控步骤能垒只有85.6 kJ/mol, 这一活化能垒与贵金属的比较接近. 我们研究的12 种铁基催化剂速控步骤能垒范围为85.6~126.4 kJ/mol, 显示了配体良好的调控催化活性能力. 我们的研究结果可为高活性CO2加氢制甲酸催化剂的开发提供理论指导和参考.
