NixCuy-B24N28 催化CO2 氧化C3H8 速控步骤的反应机理研究
2022-08-13律佳媛任瑞鹏吕永康
律佳媛, 任瑞鹏, 吕永康
(1. 太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室, 山西 太原 030024;2. 山西浙大新材料与化工研究院, 山西 太原 030024)
丙烷(C3H8)催化氧化脱氢是有效利用天然气和页岩气的重要手段, 二氧化碳(CO2)作为弱氧化剂参与催化反应, 这对于能源利用和解决全球变暖问题具有重要的研究价值[1-2]. 近年来, CO2氧化C3H8反应逐渐成为当前研究的热点[3-4], 其中C3H8的第一个C-H 键活化是氧化脱氢过程的关键步骤[1,5-6]. C3H8初步氧化可以通过C(1)-H或C(2)-H 键的断裂进行, 生成的中间体可以再继续裂解.目前大部分研究表明C3H8的C(2)-H 键断裂更具优势, 而且, C3H8的C-H 键断裂生成丙基为速率控制步骤, 在C3H8氧化脱氢过程中起着决定性作用[7-9].
目前, 推动C3H8氧化脱氢的关键在于催化剂的设计. 研究表明, 氮化硼(BN)边缘能有效的吸附并选择性活化C3H8的C-H 键[10-11], 促进C3H8的氧化脱氢, 但因其大带隙[12]对CO2的吸附活化并不敏感, 而Baei[13]和Qu[14]等研究发现BN 双环团簇可以有效地捕获并激活CO2分子. 另外,Yang 等[15-18]发现在BN 团簇内掺杂少量过渡金属可以调节B-p轨道的占据, 在催化反应中表现出良好的催化活性. 研究表明, 相对于纯的BN 材料, 有金属颗粒掺杂的BN 类催化体系展示出相对更好的催化性能, 将其用于CO2氧化C3H8反应, 或可同时促进C3H8的C-H 键断裂脱氢与CO2的活化加氢.
在目前广泛研究的C3H8脱氢催化剂中, 镍基催化剂可作为C3H8氧化脱氢的有效催化剂, 如介孔NiO 在450 ℃时, 催化C3H8氧化的转化率可达25.8%, C3H6选择性为51.3%, 且72 h 内保持稳定[19],但镍基催化剂的NiOx易被还原, 促进C3H8的CC 键断裂同时抑制C-H 的活化[20]. 双金属合金相对于孤立的金属基催化剂在催化C3H8选择性脱氢方面则具有更为优异的C-H 活化性能, 如Ni/Mo 摩尔比为3/1 的NiMo/Al2O3催化剂在570 ℃下的C3H6选择性为63.1%[21]; Fe3Ni(111)表面亦更倾向于C3H8的C-H 键氧化裂解[22]; PtCu 合金催化剂在催化丙烷直接脱氢(PDH)过程中, C3H8的C-C 键断裂的能垒(2.0 eV)远高于C-H 键(1.54 eV), 表明该体系更利于C-H 键活化[23]. Cu 作为Ni 的邻位金属, 具有与其相似的电子分布状态, 且Cu 基催化剂在CO2活化方面有着不可替代的优势,Ni-Cu 双金属间的相互作用或能改善活性中心的化学环境, 进而显著提高催化剂的活性[24]. 因此, 为了同时活化C3H8和CO2, 助力C3H8氧化脱氢, NiCu双金属合金催化体系或可作为一种不错的选择. 但Ni 和Cu 两种金属的含量占比作为影响催化性能的重要因素, 仍有待探究.
在这项工作中, 所涉及到的催化剂模型均为双金属共掺杂的硼氮类富勒烯. 利用VASP 软件理论计算了C3H8和CO2在催化剂NixCuy-B24N28(x+y=4,x=1、 2、 3、 4)表面相应的吸附能和反应活化能, 并探讨了CO2氧化C3H8反应速控步骤的吸附以及氧化机理.
1 计算细节及催化剂构型
1.1 计算细节
DFT 计算均使用Vienna Ab-initio Simulation Package 模拟软件包(VASP)进行[25-27], 所用模型为非周期性簇模型, 同时在Z 轴方向设置1.5 nm 的真空度, 构型优化过程中所有原子均处于驰豫状态且不考虑自旋极化. 运用投影仪增强波(PAW)[28]描述离子核与电子之间的相互作用, 通过广义梯度近似(GGA)[29]处理交换相关函数, 截断能设定为270 eV, 布里渊区k 点采样为1×1×1[30]. 采用CINEB 方法[31]确定过渡态(TS), 之后进一步计算虚频, 有且仅有一个虚频证实过渡态计算正确.
吸附能定义如下:
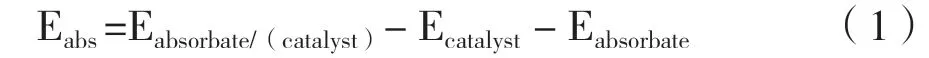
其中Eabsorbate/(catalyst)、 Ecatalyst、 Eabsorbate分别意味着吸附物种吸附在催化剂上的总能量、 催化剂本身的能量以及吸附物种本身的能量.
对于在催化剂NixCuy-B24N28表面上发生的A+B →C+D 之类的反应, 通过以下公式计算反应热(ΔH)和活化能垒(Ea):
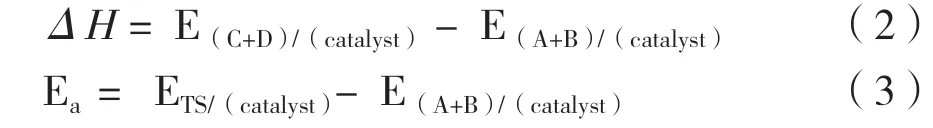
其中, E(A+B)/(catalyst)表示催化剂表面共吸附的反应物即A 和B 的总能量, E(C+D)/(catalyst)表示产物即被吸附的C 和D 的总能量, ETS/(catalyst)是反应过渡态的能量.
p 带中心的公式如下所示[32]:

其中, E 代表能量, P(E)代表p 电子密度.
1.2 催化剂构型
我们所用催化剂为双金属共掺杂的硼氮类富勒烯, 这是一个(BN)28笼状模型, 通过电弧熔炼合成在其顶部以4 个金属原子取代掺杂4 个B 原子的结构[33-35]. 以NiCu3-B24N28为例, 模型的主视图和俯视图如图1 所示. 深蓝色为氮原子, 粉色为硼原子, 黄色为铜原子, 浅蓝色为镍原子.

图1 NiCu3-B24N28催化剂的主视图和俯视图Fig.1 Front view and top view of the NiCu3-B24N28 catalyst
2 结果与讨论
2.1 C3H8初步氧化的物种在NixCuy-B24N28表面的吸附
(BN)28富勒烯是由28 个硼原子和28 个氮原子组成的四元环与六元环依次相连的稳定笼结构. 我们计算了金属于不同位点取代掺杂(BN)28簇模型的形成能, 搭建了如图1 所示的以4 个金属粒子取代4 个B 原子的稳定催化剂模型, 之后确定了C3H8、 CO2及其中间体的吸附态, 包括CH3CH2CH3、CH2CH2CH3、 CH3CHCH3、 OCO 、 OCHO 和HOCO. 各物种在NiCu3-B24N28表面的稳定吸附构型图如图2所示, 而在其它NixCuy-B24N28表面的吸附构型与在NiCu3-B24N28表面类似, 与之对应的吸附能列于表1.
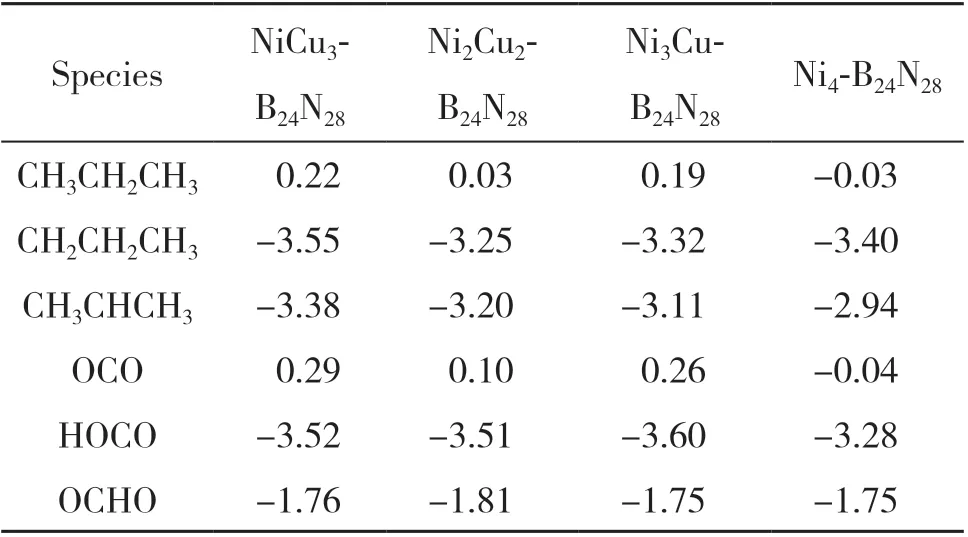
表1 C3H8及相关物种在NixCuy-B24N28表面的吸附能(eV)Table 1 Adsorption energy(eV) of C3H8 and related species on the surface of NixCuy-B24N28

图2 C3H8及相关物种在NiCu3-B24N28表面的稳定构型图Fig.2 The stable configuration of C3H8 and related species on NiCu3-B24N28 surface
C3Hx(x= 7~8)的 吸 附: C3H8吸 附 在H1 位点的上方与Cu-N-Cu 组成的三元环相互作用, 其在NixCuy-B24N28表面吸附最稳定的结构是C-C键, 几乎与催化剂表面的Cu-N 键平行, 且为物理吸附. CH2CH2CH3通过C3H8伯碳的C-H 键断裂, 它的最稳定吸附状态是通过伯碳吸附在T1 位点, 通过表1 可以看出, 由于失去一个H, 其在催化剂表面的吸附能比C3H8大得多, 是化学吸附.CH3CHCH3由C3H8仲碳的C-H 键断裂生成, 通过仲碳吸附在催化剂表面的T1 位点, CH3CHCH3与CH2CH2CH3类似, 都是化学吸附.
HxCO2(x= 0~1)的吸附: CO2吸附在T2 位点的上方, 其在NixCuy-B24N28表面吸附最稳定的结构是CO2的C 原子位于T1 位点N 原子的上方, 且整个CO2分子与催化剂表面呈现15°到45°的倾斜角度, 是物理吸附. OCO 的C 原子被C3H8脱掉的H 原子攻击生成OCHO 中间体, 通过O 原子与催化剂表面N 原子之间的相互作用, 化学吸附在NixCuy-B24N28表面. OCO 通过与C3H8脱掉的H 原子结合生成HOCO, 中间体HOCO 与OCHO 类似, 都是化学吸附.
2.2 C3H8和CO2在NixCuy-B24N28表面的初步氧化过程
利用VASP 软件计算了NixCuy-B24N28表面CO2氧化C3H8反应速控步骤的反应热和活化能. 在NiCu3-B24N28催化剂表面, CO2氧化C3H8反应速控步骤的初态(IS)、 过渡态(TS)、 终态(FS)的构型如图3 所示. 其它NixCuy-B24N28表面与NiCu3-B24N28的IS、 TS、 FS 的构型类似, 相应的反应热和活化能的计算结果在表2列出.
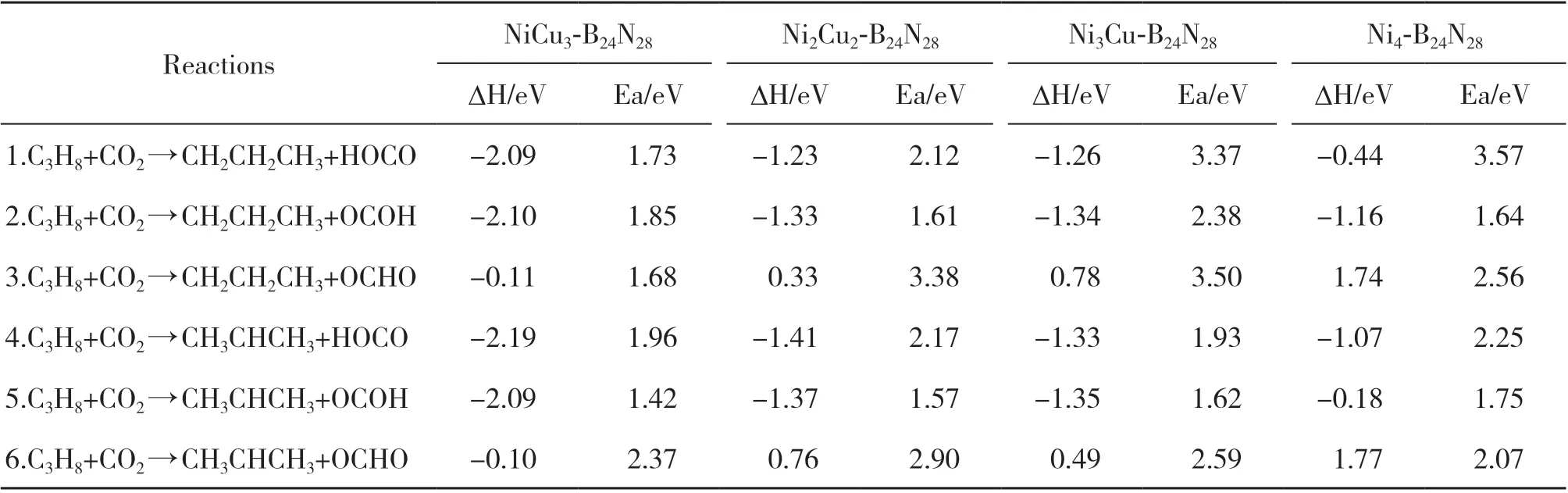
表2 NixCuy-B24N28表面CO2氧化C3H8速控步骤的相关反应热和活化能Table 2 Related reaction heat and activation energy of the rate-controlling step of the oxidation of C3H8 by CO2 on the surface of NixCuy-B24N28

图3 NiCu3-B24N28表面CO2氧化C3H8速控步骤的IS、 TS、 FS的稳定构型图Fig.3 The stable configuration of IS, TS and FS of the rate-controlling step of the oxidation of C3H8 by CO2 on the NiCu3-B24N28 surface
R1: C3H8+CO2→CH2CH2CH3+HOCO. C3H8在NiCu3-B24N28表面的初始氧化始于C3H8和CO2分子的吸附. C3H8的C-C键与表面平行, CO2与表面呈45°倾斜角, 作为反应物的初始构型. C3H8的初始氧化有六条路径, 分别是C(1)-O(1)、 C(1)-O(2)、C(1)-C、 C(2)-O(1)、 C(2)-O(2) 和C(2)-C键结合路径. 该反应表示的是C3H8和CO2开始初步氧化,经过C3H8伯碳脱氢与CO2的O结合, 即通过C(1)-O(1)路径生成CH2CH2CH3和HOCO. CH2CH2CH3和HOCO通过C原子分别与催化剂表面的T1、 T2 位点成键, 该反应放热-2.09 eV, 活化能为1.73 eV.
R2: C3H8+CO2→CH2CH2CH3+OCOH. C3H8伯 碳脱氢也可以和CO2的另一个活性O结合, 即通过C(1)-O(2)路径生成CH2CH2CH3和OCOH, 该反应放热-2.10 eV, 需克服1.85 eV的能垒. 其活化能相对于C(1)-O(1)路径较高, 因此不支持该路径优先反应.
R3: C3H8+CO2→CH2CH2CH3+OCHO. C3H8伯 碳脱氢还可以与CO2的C结合, 即通过C(1)-C路径跨越1.68 eV的能垒, 生成CH2CH2CH3和OCHO. 可见C(1)-C路径所需的活化能略低于C(1)-O(1)路径, 两者之间存在较激烈的竞争(1.68、 1.73 eV), 但前者较容易.
R4: C3H8+CO2→CH3CHCH3+HOCO. C3H8还可以通过仲碳脱氢与氧化剂CO2的O结合, 该过程中反应放热为-2.19 eV, H原子需克服1.96 eV的能垒, 通过C(2)-O(1)路径生成CH3CHCH3和HOCO. 将其与伯碳脱氢的3条路径对比, 发现R4路径发生反应的概率较低.
R5: C3H8+CO2→CH3CHCH3+OCOH. C3H8通 过仲碳脱氢经TS5形成CH3CHCH3和OCOH的活化能为1.42 eV, 反应放热-2.09 eV. 根据表2中的活化能数据, 可以发现在NiCu3-B24N28表面, C(2)-O(2)路径反应所需活化能最低, 且低于同路径下的Ni2Cu2-B24N28、 Ni3Cu-B24N28以及Ni4-B24N28表面, 因此可作为最有可能发生的路径, 这与之前的研究结果一致[36].
R6:C3H8+CO2→CH3CHCH3+OCHO. C3H8仲 碳脱氢的C(2)-C路径也可能发生, 该过程放热-0.10 eV, 活化能为2.37 eV. 生成CH3CHCH3和OCHO的活化能高于生成CH3CHCH3和OCOH的活化能, 说明C(2)-O(2)路径更有利.
通过图4 可以发现, NixCuy-B24N28催化CO2氧化C3H8反应中C3H8的第一个C-H键断裂的活化能随着Cu占比的增大逐渐降低, 这说明催化活性与催化体系中掺杂金属原子的相对数量有一定的标度关系. 在NixCuy-B24N28催化剂表面, C3H8的C-H键的活化以及CO2的C-O键的活化可能与Cu驱使的双金属向BN层的电子转移数量有关.
2.3 讨论
我们计算了NixCuy-B24N28表面C(2)-O(2)路径中丙烷C(2)原子及催化剂N 原子的p 轨道的局域态密度(PDOS)及其p 带中心, 图5 中0 点为费米能级, 不同颜色的曲线分别代表C 原子(黑色)和N 原子(红色)的PDOS, 垂直实线分别表示其p 带中心. 由图5(a)-(d)可知, 在双金属掺杂催化体系中, C3H7中间体C 原子与吸附位点N 原子的p 轨道的重叠程度逐渐增大, 即C 原子与N 原子p带中心差值分别为NiCu3-B24N28(0.52 eV)、 Ni2Cu2-B24N28(0.82 eV)、 Ni3Cu-B24N28(0.96 eV) 和Ni4-B24N28(1.06 eV). 结果表明, 随着Cu 含量占比的逐渐升高, 丙烷C 原子与催化剂表面N 原子之间的电子转移量逐渐增大, 使得催化剂对丙烷C-H键的活化作用逐渐增强, 反应所需的活化能逐渐降低. 该分析结果与我们所得活化能变化规律相匹配.

图5 NixCuy-B24N28表面C原子与N原子p 轨道的PDOS及p 带中心Fig.5 The PDOS and p-band center of p orbital of carbon atom and nitrogen atom on the surface of NixCuy-B24N28
3 结论
通过VASP 软件计算了CO2氧化C3H8速控步骤中6 条可能路径下的反应热和活化能, 研究了C3H8和CO2在NixCuy-B24N28(x+y=4,x=1、 2、 3、 4)催化剂表面吸附和速控步骤的反应机理. 根据计算结果得出以下结论: (1) C3H8和CO2在NixCuy-B24N28表面是物理吸附, C3H8+CO2→CH3CHCH3+OCOH是最有利的路径. (2)在NixCuy-B24N28(x+y=4,x=1、2、 3、 4)催化体系中, Cu 含量的增加有助于提高CO2氧化C3H8速控步骤的活性, 其在不同催化剂表面的活化能顺序是NiCu3-B24N28(1.42 eV)、 Ni2Cu2-B24N28(1.57 eV)、 Ni3Cu-B24N28(1.62 eV)、 Ni4-B24N28(1.75 eV).
